Article Text

Abstract
Objective Non-alcoholic steatohepatitis (NASH) can progress to fibrosis, cirrhosis and end-stage liver disease. Glucagon-like peptide 1 receptor (GLP-1R) mediates β cell function. Its receptor agonists, currently used to treat type 2 diabetes mellitus, might be effective against NASH. GLP-1R, a G protein-coupled receptor family member, preferentially localises to caveolae. Therefore, we ascertained the cellular localisation of GLP-1R and caveolin (CAV)-1 in NASH liver.
Methods Liver biopsies were obtained from three patients with NASH and were compared with those of four normal patients. Immunohistochemistry (IHC) and immunoelectron microscopy (IEM) were used to compare GLP-1R and CAV-1 expression in the livers of patients with metastatic liver cancer and normal patients.
Results IHC showed that GLP-1R localised to basolateral membranes of hepatocytes with macrovesicular steatosis and was expressed in monocytes infiltrating hepatic sinusoids. CAV-1 was minimally associated with low-electron density lipid droplets (LDs) in hepatocytes. IEM showed small clusters of GLP-1R molecules on the peripheral rims of LDs and on cytoplasmic leaflets of endoplasmic reticulum membranes and vesicles, whereas CAV-1 molecules were found in LD caveolae.
Conclusions GLP-1R is present in the lipid microdomains of hepatocytes with macrovesicular steatosis. These results may help inform future studies about the liver-specific mechanisms of GLP-1 modulation in NASH therapy.
- hepatitis
- nonalcoholic steatohepatitis
- liver immunology
- liver
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Non-alcoholic fatty liver disease (NAFLD) has become the most common cause of chronic liver disease worldwide.1 In recent years, the prevalence of NAFLD has continued to rise. Approximately 10%–25% of NAFLD cases will progress to non-alcoholic steatohepatitis (NASH), whereas 10%–15% will develop to hepatocellular carcinoma.2 Hepatic lipid accumulation in NAFLD is associated with insulin resistance, oxidative stress, endoplasmic reticulum (ER) stress and mitochondrial dysfunction. Insulin resistance is characterised by impaired insulin signalling, which contributes to abnormal hepatic metabolism. It increases dysregulated gluconeogenesis by approximately twofold, a typical feature of type 2 diabetes mellitus (DM).3 Recent basic and translational studies have provided insights into the mechanisms involved in NAFLD progression. Other studies have demonstrated that macrophage-mediated inflammation plays a major role.4
Glucagon-like peptide-1 (GLP-1), a 30-amino acid peptide secreted by Langerhans cells, has an antihyperglycaemic effect by stimulating insulin secretion and reducing glucose-dependent glucagon secretion.5 Whether GLP-1 secretion is lower in patients with type 2 DM remains controversial.6 The GLP-l receptor (GLP-1R) is expressed in the islet cells, lung, brain, kidney and adipose tissue of animals and humans.7 The receptor is a member of the class B family of G protein-coupled receptors (GPCRs), which are characterised by their ability to bind caveolin (CAV)-1, a 21–24 kDa protein that is the principal protein component of lipid raft/caveolae microdomains of plasma membranes.8 9 In experimental NAFLD models, CAV-1 is expressed on the plasma membrane and in the cytoplasm, including mitochondria, ER and membranes of lipid droplets (LDs).10
GLP-1R expression has been shown not only in the hepatocyte cell lines HuH7 and HepG2 but also in primary human hepatocytes in vitro.11 Nevertheless, few reports have described the distribution of GLP-1R in intact human liver. Therefore, we aimed to characterise the distribution and subcellular localisation of the GLP-1R and CAV-1 in human NASH liver samples using immunohistochemistry (IHC) and immunoelectron microscopy (IEM).
Materials and methods
Antibodies
GLP-1 antibody was obtained from Abcam (Cambridge Science Park, Cambridge, UK). CAV-1 antibody was from BD Biosciences (Franklin Lakes, New Jersey, USA).
Materials
The results of liver needle biopsies of three patients with early stage NASH were compared with those of four patients with normal livers and metastatic liver cancer. Additional details are provided in online supplementary materials and methods.
Supplemental material
Methods
Details of the IHC and IEM on the biopsies are provided in online supplementary materials and methods.
Results
NASH biopsy specimens were obtained from patients with clinically coexisting metabolic conditions, including type 2 DM and hyperlipidaemia, but not from alcohol consumers.
In normal liver, a low level of GLP-1R expression was observed in a few sinusoidal cells (figure 1A,B). CAV-1 was expressed in vessel walls, including those of the portal vein, and in the cells lining the sinusoids in zone 3 of normal liver specimens (figure 1C,D).

Immunohistochemistry and western blot analysis of glucagon-like peptide 1 receptor (GLP-1R) and caveolin-1 (CAV-1) in serial sections of normal and non-alcoholic steatohepatitis (NASH) liver. (A, B) Expression of GLP-1R (brown) in normal liver. A few cells showed low expression of GLP-1R in control liver. (C, D) Expressions of CAV-1 (brown) in normal liver. CAV-1 is expressed in the hepatic artery, capillary venule and portal vein in the portal tract (P), and in the cells lining the hepatic sinusoids around pericentral zone 3. The left and right panels show low-magnification and high-magnification images, respectively, of the pericentral region. (A, C) Low magnification (×100). (B, D) High magnification (×400). (E–H) Expression of GLP-1R and CAV-1 in NASH liver. (E) Expression of GLP-1R was observed in hepatocytes and inflammatory cells. (F) Most GLP-1R-positive cells were hepatocytes at the basolateral membrane with macrovesicular steatosis and mononuclear cells. (G) CAV-1 expression is evident mainly in the sinusoidal lining cells. (H) CAV-1 expression in hepatocyte lipid droplets (LDs). Left and right panels show low-magnification and high-magnification images of the central vein, respectively. (E, G) Low magnification (×100). (F, H) High magnification (×400). White arrows indicate the sinusoidal lining cells and black arrows indicate the LDs of macrovesicular cells. Black arrowheads indicate the hepatocyte basolateral membrane and white arrowheads indicate mononuclear cells. P denotes the portal tract; C denotes the central vein. (I) Western blot analysis of GLP-1R expression in control and NASH liver lysates. Lysates containing 30 µg protein were subjected to Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and analysed by western blotting. Lanes 1–3: normal liver; lanes 4 and 5: NASH liver. GLP-1R protein expression was significantly higher in NASH.
In NASH livers, as presented in figure 1C,D, GLP-1R expression is visible in cells lining the sinusoids in areas with steatosis and features of inflammation (figure 1E,F). More specifically, GLP-1R immunoreactivity is mainly visible in inflammatory cells, basolateral hepatocytes and areas of macrovesicular steatosis with mononuclear cells (figure 1F). Furthermore, CAV-1 immunoreactivity in the cells lining the hepatic sinusoids was more intense in biopsies from patients with NASH (figure 1G,H). Moreover, immunoreactive CAV-1 was present in the LDs of hepatocytes, with a low level of expression in infiltrating monocytes/macrophages (figure 1H).
In an attempt to corroborate the IHC findings, we used western blotting to measure GLP-1R protein expression in control liver tissues and in liver tissues with NASH. The results confirmed that GLP-1R expression was low in control samples and high in NASH samples (figure 1I).
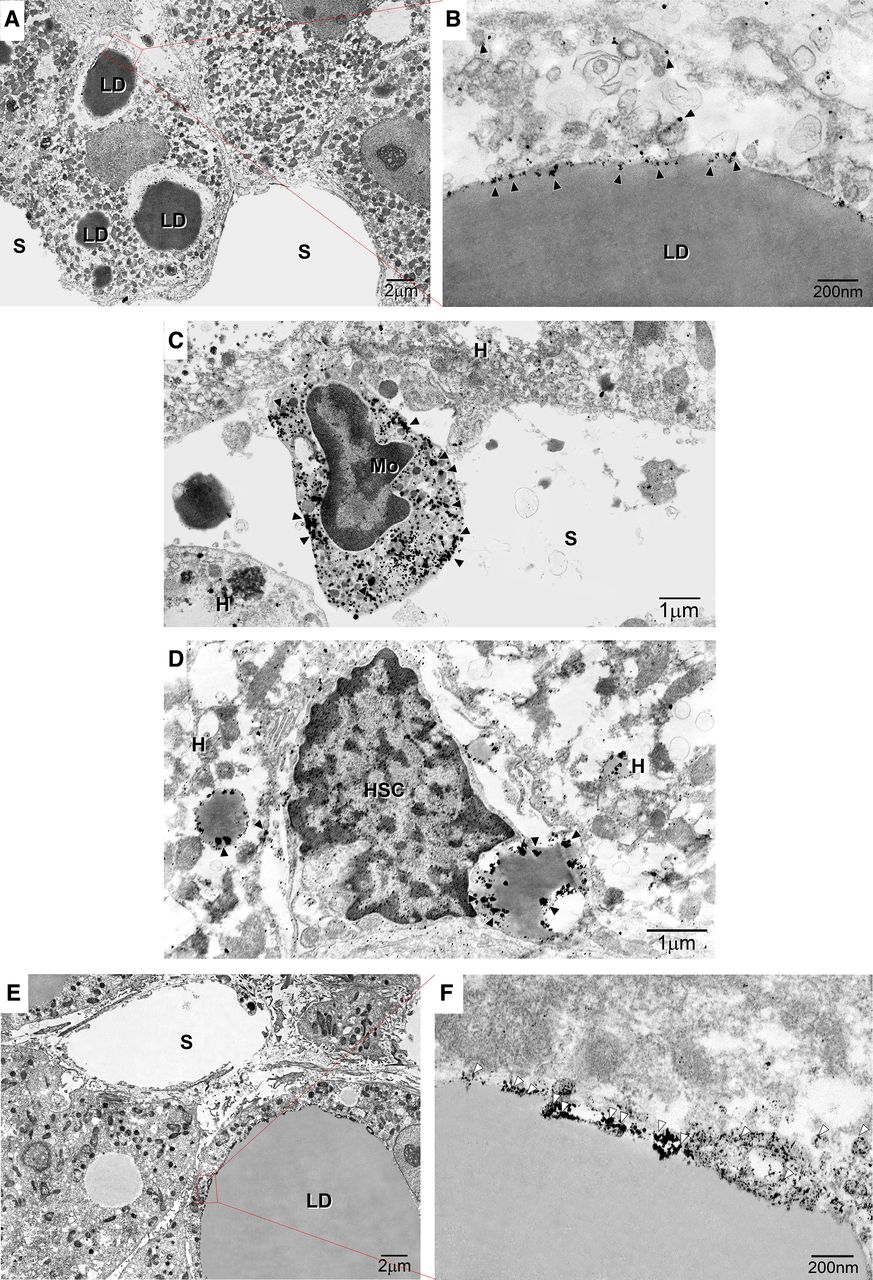
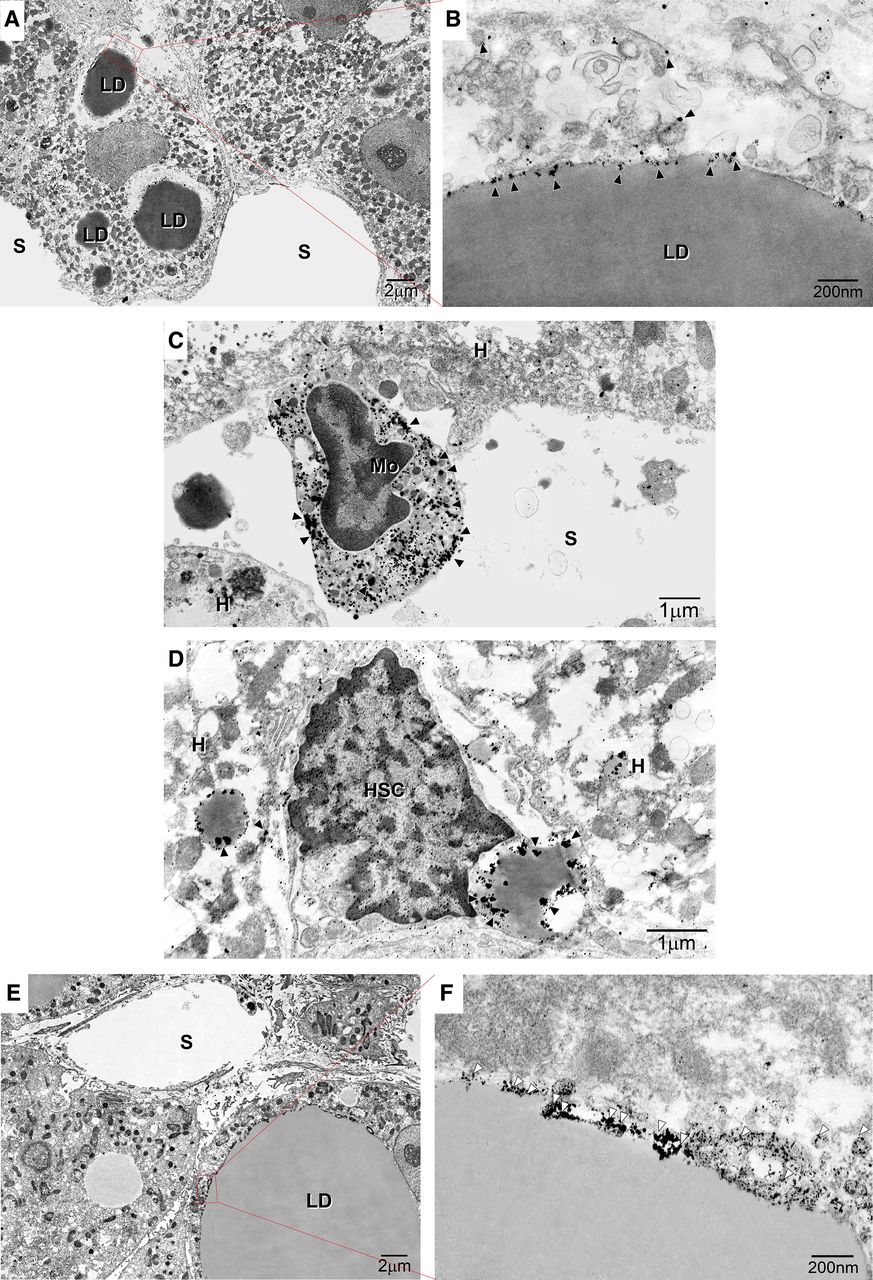
IEM revealed gold-labelled GLP-1R particles mostly within monocytes/macrophages (figure 2A,B) and the caveolae of LDs, ER and vesicles in hepatocytes (figure 2A,B). Moreover, GLP-1R expression was detected in the LDs of hepatic stellate cells (figure 2D) and monocytes/macrophages (figure 2E). CAV-1 immunolabelling was observed in small clusters on low-electron density LDs in macrovesicular hepatocytes (figure 2F,G).
{kind=link}
{kind=link}
Immunoelectron microscopy of the subcellular localisation of glucagon-like peptide 1 receptor (GLP-1R) and caveolin-1 (CAV-1) in non-alcoholic steatohepatitis (NASH) liver. (A) Image showing marked macrovesicular steatosis: large lipid droplets (LDs) are visible in hepatocytes. (B) Immunogold particle labelling of GLP-1R on endoplasmic reticulum (ER)-like membranes attached to LDs and vesicles around LDs. (C) GLP-1R is localised to the peripheral rims of LDs in hepatic stellate cells and LDs in hepatocytes. (D) GLP-1R is localised to the plasma membrane and organelles of monocytes on hepatic sinusoids. (E) Macrovesicular and microvesicular LDs are visible in hepatocytes. (F) Immunogold particle labelling of CAV-1: CAV-1 is expressed on the caveolae of LDs. White arrowheads indicate CAV-1. A continuous basal lamina is apparent underneath the endothelium. Magnification=×20 000. Bar=200 nm. HSC, hepatic stellate cell. Arrowheads indicate CAV-1-positive particles. Black arrowheads indicate caveolae. Black arrows indicate LD caveolae. White arrows indicate ER-like membranes. Magnification=×2000. Bar=1 µm or 200 nm. White arrowheads indicate GLP-1R or CAV-1. Bar=200 nm. HSC, hepatic stellate cell. Black arrowheads indicate caveolae. Black arrows indicate LD caveolae. K denotes Kupffer cells. LD denotes lipid droplets.
Discussion
Using IHC and IEM, we have ascertained the distribution and subcellular localisation of GLP-1R in liver biopsies from patients with NASH. Pyke et al tested the available rabbit polyclonal Abs that had been recommended for the identification of human GLP-1R by IHC using paraffin-embedded material, but there has been little or no validation of their findings.7 However, we used a more specific mouse monoclonal antibody (Mab) against the human GLP-1R extracellular domain and found it to be more effective for IHC in several paraffin-embedded tissues.7 We detected weak expression of GLP-1R using the earlier antibody and this MAb in normal liver. However, a higher level of GLP-1R expression was found in NASH samples. This expression was localised to LDs on the basolateral membranes of hepatocytes with macrovesicular steatosis and infiltrating mononuclear cells. Moreover, IEM revealed that GLP-1R molecules formed small clusters on the rims of LDs and that they were present in the cytoplasmic leaflets of ER membranes and vesicles of basolateral hepatocyte membranes.
The ‘multiple-parallel hit’ model of NASH pathogenesis posits that three factors (environmental, metabolic and genetic factors) contribute to the development and progression of NASH by affecting diverse organs, such as the liver, intestine and adipose tissue.12 Excessive lipid accumulation in the liver causes hepatocellular lipotoxicity by inducing cellular and organelle oxidative stress, even in the ER, and mitochondrial dysfunction, which eventually leads to hepatocyte cell death.12 In addition, chronic ER stress results in hepatic lipid accumulation by activating de novo fatty acid synthesis,12 suggesting that a vicious cycle involving ER stress and hepatic steatosis is involved in the development and progression of NAFLD/NASH. Liver macrophages make up the largest proportion of tissue macrophages in the host and consist of two dissimilar groups: Kupffer cells (KCs) and monocyte-derived macrophages. Macrophage polarisation has been classically clustered into two major macrophage polarisation programmes, classically activated macrophages or M1 and alternatively activated macrophages or M2, each related to specific immune responses, among which both progression and resolution of inflammation constitutes a critical determinant. As previously reported, GLP-1R stimulation increased foam cell formation in monocytes and interleukin-6 (IL-6), tumour necrosis factor-α and IL-1β production in obese patients.13
IHC showed that GLP-1R expression was localised mainly to the basolateral membranes of hepatocytes and LDs and to monocytes/macrophages. However, CAV-1 expression was localised to LDs in hepatocytes and showed limited expression in infiltrating mononuclear cells. The expression of numerous GPCRs has been observed in membrane rafts/caveolae. Diseases associated with aberrant signalling can result in altered localisation or expression of signalling proteins in caveolae.8 9 Furthermore, overexpression of a dominant-negative form of CAV-1 or mutations within the CAV-1-binding domain of the GLP-1R attenuates GLP-1 binding and GLP-1R expression at the membrane.14 The segregation of caveolae from LDs has been confirmed visually in steatotic macrovesicular hepatocytes, in which CAV-1 was spatially separated from GLP1-R.15 Consequently, CAV-1 might play a role in maintaining the balance of these signalling molecules.8 Moreover, the ectopic expression of CAV-1 was protective against fatty acid-mediated lipotoxicity in hepatocytes.9 Studies to address this important functional abnormality in injured hepatocytes are expected to be underway.
In conclusion, our findings emphasise the importance of additional experimentation to identify the mechanisms and conditions linking GLP-1R activation and CAV-1 to the pathophysiology of hepatocyte lipid metabolism and ectopic lipid deposition in NASH. These results may inform future studies, and this work may have implications for our understanding of the mechanisms of GLP-1 agonists in the treatment of NASH.
Acknowledgments
The authors would like to thank Hitoshi Yamazaki and Yoshihito Takahashi of Kitasato University Medical Center for providing liver samples. The authors also thank Mariko Ogi, Tomoko Yoshii and Natsuki Hata for their technical assistance.
References
Footnotes
Contributors HY and WA designed the study. HY and WA conducted the experiments. HY, WA and MO wrote the manuscript.
Funding HY received scholarship support from AbbVie GK, Gilead Sciences, Merck Sharp & Dohme, Astellas Pharma, Otsuka Pharmaceutical, Zeria Pharmaceutical, EA Pharma, Shionogi and Co. Ltd. and Chugai Pharmaceutical. None of the study sponsor(s) had any role in the study design, in the collection, analysis and interpretation of the data; in the writing of the report; or in the decision to submit the paper for publication.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement No data available. All data in this article.