Article Text

Abstract
Introduction Population-level screening has been shown to reduce the incidence and mortality of colorectal cancer (CRC). Unfortunately, adherence to screening recommendations among eligible US adults remains below national goals. A relatively new non-invasive screening modality, the Food and Drug Administration–approved multi-target stool DNA (mt-sDNA) assay (commercialised as Cologuard), which combines the detection of haemoglobin and DNA abnormalities, has been completed by more than 3 million individuals. Given mt-sDNA’s recent availability, the effectiveness of mt-sDNA screening with respect to CRC incidence and mortality reduction has not yet been established.
Methods and analysis Through an academic–industry collaboration, a prospective cohort study (Voyage) was designed with an initial enrolment target of 150 000 individuals with mt-sDNA ordered by their healthcare provider for CRC screening. Consented participants will be asked to complete a baseline questionnaire to collect sociodemographic and health information. Additional questionnaires will be provided after 1 year, and every 3 years thereafter, to collect data regarding CRC screening follow-up in order to estimate rates of CRC incidence and other health outcomes. Linkage to the National Death Index will be used to estimate mortality rates.
Ethics and dissemination The Voyage study will be conducted in accordance with international guidelines and local regulatory requirements and laws. Data will be stored and retained at Mayo Clinic. Only limited data elements required for research purposes will be transmitted between Mayo Clinic and Exact Sciences Laboratories. Results of the Voyage study will be disseminated through scientific presentations and publications.
Trial registration number NCT04124406.
- cancer prevention
- colorectal cancer screening
- colorectal cancer
- clinical trials
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Colorectal cancer (CRC) is the second deadliest cancer, with an estimated 145 600 new cases and 51 020 deaths in the USA in 2019.1 An effective strategy to reduce CRC incidence and improve overall survival is regular CRC screening.2 Despite the availability of multiple screening tests to improve early detection of CRC, nearly one-third of eligible US adults have never been screened.3 Current CRC screening rates fall below the National Colorectal Cancer Roundtable goal of 80% adherence among eligible adults.4 Commonly identified barriers to CRC screening include (1) limited usage of tools to remind screening-eligible patients by providers, (2) a large number of patients who do not regularly see a healthcare provider, (3) a focus on invasive screening methods with high patient burden, (4) a lack of patient awareness of screening options and recommendations, and (5) socioeconomic challenges such as financial barriers and limited healthcare access.5 6
The multi-target stool DNA (mt-sDNA) assay (commercialised as Cologuard; Exact Sciences, Madison, WI) has contributed to an increase in population-wide adherence to CRC screening among average-risk individuals.7 Major national healthcare organisations and guideline committees recommend mt-sDNA screening at 3-year intervals.2 8–10 Mt-sDNA combines the detection of occult haemoglobin using a immunochemical assay with quantitative detection of KRAS mutations, aberrant NDRG4 and BMP3 methylation, and ACTB.11 Compared with a leading faecal immunochemical test (FIT, OC FIT-CHEK; Polymedco), mt-sDNA demonstrated significantly improved sensitivity for detecting cancer and precancer in a large, cross-sectional, multi-centre study.11 Unlike colonoscopy, sigmoidoscopy or other screening modalities that require a separate clinical encounter and bowel preparation, mt-sDNA is non-invasive and is completed at home with a specialised stool sample collection kit.2 As of October 2019, more than 3 million people have been screened with mt-sDNA since it was approved by the Food and Drug Administration (FDA) in 2014.12
Although performance characteristics of mt-sDNA have been established in a prospective study of nearly 10 000 participants,11 the real-world impact of mt-sDNA on long-term clinical outcomes, such as CRC incidence and mortality, has not been determined. To address this knowledge gap, we designed a large-scale, nationwide cohort study (Voyage) to examine CRC-related patient use of healthcare services and outcomes over time, including incidence and mortality endpoints. The Voyage study will be conducted in an anticipated cohort of 150 000 participants who had mt-sDNA for CRC screening ordered by their healthcare provider. Voyage is a collaboration between Mayo Clinic and Exact Sciences.
Methods and analysis
Study design
Using a prospective cohort study design, Voyage will recruit individuals who received mt-sDNA for CRC screening that was ordered by their healthcare provider. Our initial enrolment target is 150 000 participants. Individuals will be selected for recruitment via a random sample of individuals with a mt-sDNA order that was received and validated by Exact Sciences Laboratories (ESL). Individuals must live within the 50 states in the USA or Puerto Rico. After providing informed consent, participants will be followed prospectively for healthcare utilisation and health outcomes, which will be evaluated through periodic contacts. Participants will be asked to complete a baseline health questionnaire (T0), a 1-year follow-up questionnaire (T1) and additional follow-up questionnaires every 3 years thereafter.
Data will be obtained from medical records to validate patient-reported outcomes for participants who provide medical record release. Linkage to the National Death Index will be used to ascertain participant deaths not reported to the study. Process and participant documents related to the Voyage study were reviewed and approved by members of the Mayo Clinic Health Research Advisory Council.
The mt-sDNA testing and reporting process will remain unchanged for participants in the Voyage study. Mt-sDNA test results will be reported qualitatively as positive or negative, which corresponds to its FDA-approved test indication. For individuals with positive mt-sDNA test results, their healthcare providers will receive a Patient Report that suggests a follow-up diagnostic colonoscopy, which is in accordance with recommendations from major guideline organisations.2 8 10
Assuming a 3-year enrolment period, Voyage is expected to continue for 7 years following the date of last patient enrolment, with the intention of following the cohort to evaluate longer-term outcomes. Seven years of participant follow-up after enrolment would allow for an estimate of the mt-sDNA rescreening rate for two complete screening cycles (figure 1).

Timeline of the Voyage study.
Study aims
To better characterise the relationship between mt-sDNA screening and patient outcomes, we propose to complete the following study aims:
Aim 1: Enrol and collect baseline (T0) health survey data on 150 000 participants who received a mt-sDNA test order.
Aim 2: Collect and analyse healthcare utilisation and health survey data 1 year post-enrolment (T1) to (a) determine the proportion of individuals who report having received a diagnostic colonoscopy or structural examination of the colon and (b) evaluate findings from diagnostic colonoscopies after completing mt-sDNA.
Aim 3: Initiate longitudinal follow-up to determine the rates of CRC incidence and mortality among participants who completed mt-sDNA. This aim will be completed when sufficient follow-up data are available.
Aim 3A: Assess whether the age-adjusted and sex-adjusted CRC incidence rates differ from national incidence rates reported by the Surveillance, Epidemiology and End Results (SEER) registry,13 after adjusting for high-risk individuals in the national dataset. We hypothesise that the observed incidence among those screened with mt-sDNA will be less than the expected incidence (see Statistical methods section for a description of the expected comparator value).
Aim 3B: Assess whether the age-adjusted and sex-adjusted CRC mortality rates differ from national mortality rates reported by the SEER registry,13 after adjusting for high-risk individuals in the national dataset. We hypothesise that the observed CRC-related mortality among those screened with mt-sDNA will be less than the expected mortality (see Statistical methods section for a description of the expected comparator value).
Inclusion and exclusion criteria
To be eligible for participation in the Voyage study, individuals must be residents of the USA or Puerto Rico, age 18 years or older, able to provide informed consent, able to complete questionnaires in English or Spanish, and have received an order for an mt-sDNA test from an authorised healthcare provider in the USA or Puerto Rico. Those who receive a mt-sDNA order from a health system that does not allow for direct patient contact by ESL will be excluded from the enrolment process.
Participant enrolment
Collaborators at ESL will randomly select eligible participants from among all patients with an order for mt-sDNA during the enrolment period. ESL will mail a study introduction letter and response form (in both English and Spanish) to the randomly selected eligible participants. ESL will not directly provide Mayo Clinic with contact information for eligible participants. Individuals who are interested in participating will be instructed to mail the response form in an envelope provided by ESL to the study team at Mayo Clinic. Eligible participants will indicate whether they prefer to receive future study materials in English or Spanish.
The study team at Mayo Clinic will send an invitation packet to all eligible participants who return an affirmative response form. Documentation of informed consent will be obtained from participants in compliance with Good Clinical Practice (GCP) and local regulatory and legal requirements. The informed consent form used in this study, and any changes made during the study, will be prospectively approved by the Mayo Clinic Institutional Review Board (IRB) before use. Consent forms will be sent via US mail and returned in a prepaid envelope. All signed consent forms will be scanned to create an electronic copy and paper originals will be stored at Mayo Clinic.
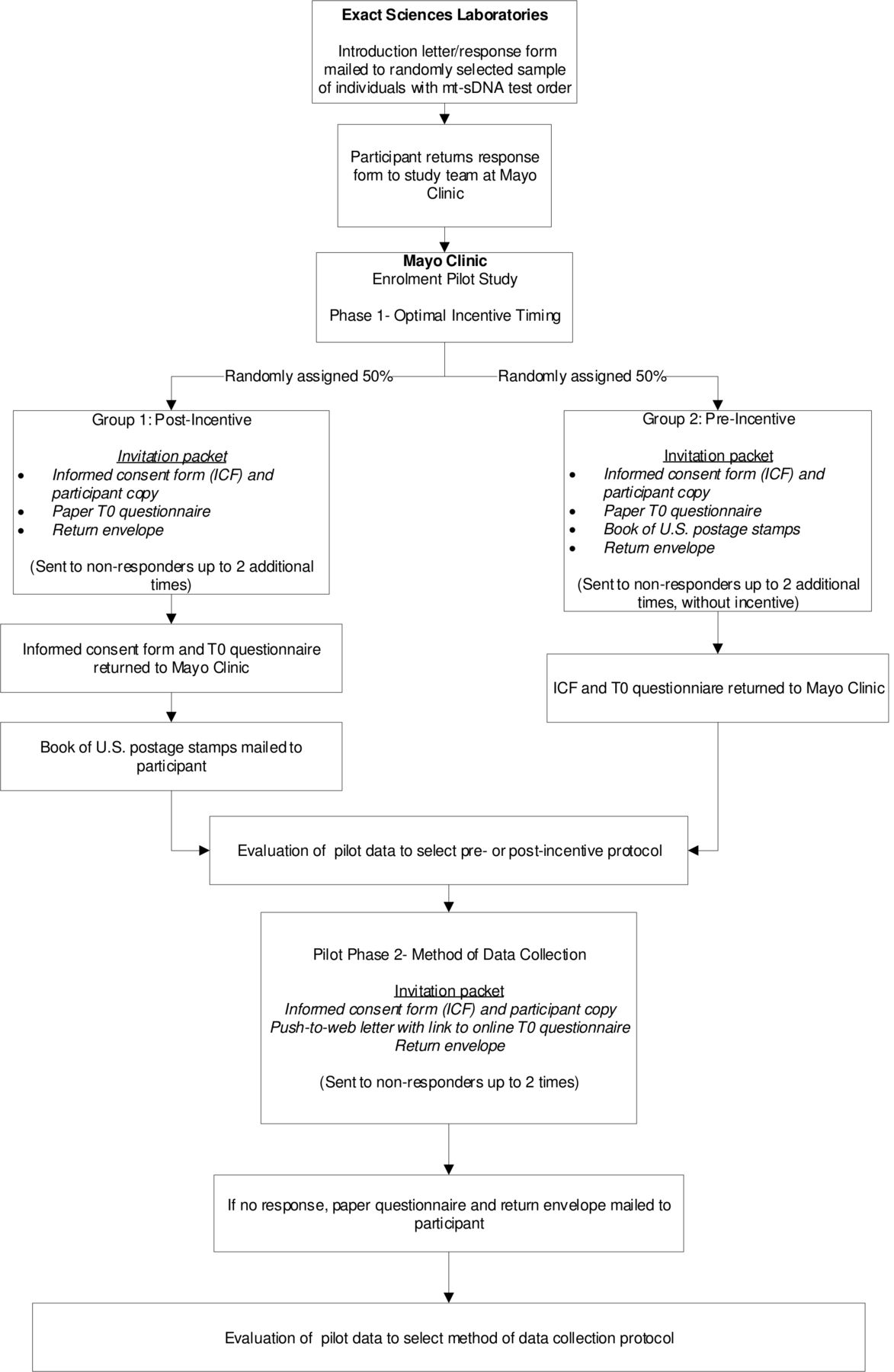
We will incentivise participant response with a book of 20 US postage stamps (current estimated value of US$11.00), similar to other Mayo Clinic prospective studies.14 To determine the optimal survey administration protocol that maximises response rates while balancing the cost per completed survey, we will conduct an enrolment pilot study in which we will vary the timing of the incentive and the method of data collection (figure 2). Phase I of the pilot study will determine the optimal timing for the study incentive. During phase I, participants will be randomly assigned to one of two groups: (1) the study incentive is sent after the T0 questionnaire is returned to Mayo Clinic (post-incentive); and (2) the study incentive is sent in the invitation packet (pre-incentive). All participants will be offered only paper questionnaires up to three times in phase I. Phase II will investigate the method of data collection and will assess a push-to-web approach.15 All participants will receive a letter by US mail with the URL for the online questionnaire up to two times. If there is no response, a paper questionnaire and return envelope will be mailed. The approaches for incentive timing and survey administration that maximise participation and enrolment (based on response rates, questionnaire completion and project costs) will be adopted and continued long term. The complete workflow for the Voyage study is described in figure 3.

Workflow for the Voyage enrolment pilot study. The pilot study will determine the optimal timing of the study incentive (pre-incentive vs post-incentive; phase I) and most effective method of data collection (paper only vs push-to-web web–paper mixed mode; phase II) based on response rates, questionnaire completion and project costs.

{kind=link}
{kind=link}
{kind=link}
Complete Voyage study workflow.
Participant engagement
An IRB-approved T0 questionnaire covering sociodemographic information, current health status,7 personal medical history, family history of CRC, factors related to colorectal screening, CRC risk factors16 17 and healthcare access will be administered electronically via the web on the participant’s computer or mobile device, or via optical mark recognition paper copy. Questions on factors related to CRC screening and healthcare access were obtained from the Health Information National Trends Survey.18 The T1 questionnaire will repeat some questions from the T0 survey, such as health status, but will focus on healthcare utilisation and new cancer diagnoses. Additional questionnaires will be developed over time, all of which will be IRB approved prior to administration. Consent for release of medical records to validate participant-reported events related to CRC screening procedures and diagnosis will be requested.
Logistical consideration
To ensure that the Voyage study is scientifically sound, fiscally responsible, and acceptable to both patients and providers, we established the following criteria under which study continuation will be reassessed:
Overall recruitment: Opt-in response rates and participant accrual (completed informed consent and T0 questionnaire) will be monitored by Mayo Clinic. Lower-than-expected rates will trigger operational review and may require changes to procedural workflows. During the recruitment years, a cost–benefit analysis will be conducted to determine the utility of continuing the study if very low (<5%) opt-in rates are observed.
Impact on patient experience: Patient experience will be monitored through adherence rates for mt-sDNA completion. A significant drop in participant test adherence among cohort invitees and/or participants will trigger an internal review leading to study re-evaluation and possible discontinuation.
Healthcare provider experience: Rates of mt-sDNA orders will be evaluated throughout the study. Unanticipated deviations from standard ordering practices of healthcare providers may also trigger study evaluation and possible discontinuation of the study.
If the study is prematurely terminated, ESL and Mayo Clinic will ensure that study participants are notified and that relevant results are appropriately reported.
Participant withdrawal
Participants may be withdrawn from the study for failing to meet the inclusion/exclusion criteria, protocol non-compliance (eg, failure to complete the initial questionnaire within 120 days of enrolment), participant withdrawal of consent, sponsor decision (see Logistical considerations section) or other withdrawal circumstances as determined by the study team. Reasons for withdrawal and the withdrawal date will be documented by Mayo Clinic.
Study outcomes
The Voyage study is designed to enrol 150 000 participants in order to study the real-world impact of mt-sDNA on CRC incidence and mortality and to establish a cohort to enable future research to address outstanding CRC screening-related questions. If enrolment proceeds as expected, we will explore the feasibility of obtaining participant consent for residual stool sample retention (after mt-sDNA has been completed) to create a biospecimen repository. The repository would facilitate additional research that requires biological samples, including multi-omic approaches and evaluating the contribution of the microbiome.
Statistical methods
All participants who provide informed consent and return the baseline (T0) health questionnaire will be included in the primary analysis cohort. The analyses for Aims 2 and 3 will be restricted to individuals with a mt-sDNA result who fulfil the FDA-approved on-label criteria for mt-sDNA (ie, average-risk, asymptomatic individuals over 45 years old) at the time of enrolment.19 The cohort of study participants may be weighted to reflect the age and sex distribution of individuals with a mt-sDNA order during the enrolment period if indicated by non-response bias analyses. Responses to the questionnaires at T0 and T1 will be summarised using standard descriptive statistics. A 95% CI will be constructed using an exact method for binomial parameter for the proportion of individuals who received a diagnostic colonoscopy within 1 year of mt-sDNA testing.
Estimating CRC incidence and CRC-related mortality
The analysis of CRC incidence and CRC-related mortality, respectively, will be performed after following the participants through the end of year 10 of the study period. Both measures will be calculated using a person-years approach, which takes into account the varying duration of follow-up for each participant based on their participation in the longitudinal questionnaires and death information obtained from the National Death Index. A participant is considered to be ‘at risk’ for an event (ie, for CRC or CRC-related death) if the event has not yet occurred and the participant is known to be alive and participating in the Voyage study.
CRC incidence, both overall and age and sex specific, will be calculated as the ratio of the number of newly detected CRC cases relative to the total ‘at risk’ person-years of all Voyage participants, and expressed as a rate per 100 000 person-years. Likewise, CRC-related mortality, both overall and age and sex specific, will be calculated as the ratio of the number of CRC-related deaths relative to the total duration of participant follow-up. The rates will be directly age and sex adjusted to the US population in 2020. SEs and corresponding 95% CIs for these rates will be calculated assuming that total person-years are fixed and that the number of events (ie, CRC cases or CRC-related deaths) follows a Poisson distribution.
Incidence and mortality comparator values
Measuring the impact of mt-sDNA on population health outcomes requires an estimate of the expected CRC incidence and mortality in a comparable population of subjects not screened by mt-sDNA, with similar age, sex and race characteristics as the Voyage participants. We will calculate these values using age-specific and sex-specific SEER rates for CRC incidence and CRC-related mortality from 2009 to 2013,13 an analytical approach similar to that from the National Polyp Study.20 The SEER rates will first be age and sex adjusted to the estimated US population in 2020 and adjusted to represent the number of average-risk individuals according to mt-sDNA test indication19 before they are applied to the number of Voyage participants still ‘at risk’ to derive the expected number of CRC cases and CRC-related deaths. Although the SEER dataset only describes a subset of the US population (~30%), it co-ordinates with other federal agencies to collect data representing 96% of the total US population, including Puerto Rico.21
Standardised incidence ratios and standardised mortality ratios (SMRs) will be generated by comparing the observed number of events in the study cohort with the expected number derived from adjusted national data. Upper 95% confidence bounds for the ratios will be constructed using the formula from Rothman and Greenland.22
Sample size justification
The target size of 150 000 participants enables the development of a robust longitudinal cohort for research related to CRC screening outcomes, as described above, and also facilitates the potential creation of stool-based biospecimen repository for future research projects. Our enrolment target is feasible based on the widespread use of mt-sDNA screening (more than 3 million people screened as of October 201912) and the planned recruitment strategy to enrol participants throughout the USA and Puerto Rico.
To estimate the frequency of expected CRC-related deaths in our population, we made the following assumptions: (1) the age-matched and sex-matched distribution of participants will mirror that of individuals with an mt-sDNA order during the first quarter of 2019; (2) study enrolment will take 3 years to complete; (3) 95% of study participants will have completed mt-sDNA screening at T0; (4) 5% of participants annually will be lost to follow-up. Estimates were calculated using the age-specific and sex-specific rates for CRC-related deaths reported by SEER for 2012–201613 and for all deaths reported by the National Vital Statistics System for 2016,23 both adjusted to remove high-risk participants (estimated at ~25% of all CRC cases24 25). In general, we hypothesise that mt-sDNA screening will result in a 25% reduction in CRC-related deaths among our study participants compared with a similar population without mt-sDNA screening. This estimated mortality reduction is similar to those from previous screening studies using flexible sigmoidoscopy and faecal occult blood test, although those studies use a different methodology for population comparison.26 Notably, these assumptions were made to facilitate power calculations and might not accurately represent the final study population.
Based on these assumptions, by the end of year 10 of the study period, we could expect ~269 CRC-related deaths to occur among 150 000 participants, based on national rates. This yields an anticipated SMR of 0.75 (202/269) with an upper 95% confidence bound of 0.84, indicating that the sample size is sufficient to yield an upper bound less than 1. If a larger proportion of the participants are lost to follow-up over time and the expected number of CRC-related deaths is 225, then the upper 95% confidence bound will be 0.85 for an SMR of 0.75.
Ethics and dissemination
Study conduct
The protocol for the study was approved on 5 August 2019 by the Mayo Clinic IRB (application no. 19-000091). The study will be conducted in accordance with GCP, the provisions specified in Title 21 Parts 11, 50, 54, 56 and 812 of the U.S. Code of Federal Regulations, the International Ethical Guidelines for Biomedical Research Involving Human Subjects,27 the Declaration of Helsinki,28 and applicable legal and regulatory requirements. The investigators understand the applicable laws, have completed and will maintain compliance with human subjects protection training, and will adhere to GCP standards and the study protocol. Regulatory authorities, the IRB or Ethics Committees (ECs), and/or ESL or its designees or partners may request access to all source documents, electronic case report forms, and other study documentation for audits or inspections, including on-site at Mayo Clinic. Investigators will allow authorised individuals to access these documents. Any amendments, modifications or protocol deviations will be submitted to the IRB/ECs for review.
Data storage, access and dissemination
Electronic systems used to capture and manage study data will allow for manual and automated checks for data quality and logical consistency (eg, appropriate ranges of acceptable values). This study will use a password-protected relational SAS database created using Mayo Clinic’s SAS Data Management System and maintained by the study’s statistical team. Access to the database will be granted by the statistical team or by the study programme coordinator, all of whom will be confirmed study personnel approved by the IRB. The database will maintain a link between personal identifying information and the participant identification code. Only individuals approved to view personal identifying information will have access to the database.
The results of this study will be presented at national meetings and conferences and published in peer-reviewed journals. The results from any subsequent studies using this patient cohort will be disseminated in a similar manner. In addition, periodic updates on the outcomes of this study will be shared with the participants themselves through an annual study newsletter.
References
Footnotes
Contributors Conceptualisation: JEO, KJY, LJFR, EJK, JLSS. Methodology: JEO, KJY, LJFR, ALW, JJL. Software: EJK, CRK. Formal analysis: ALW. Investigation: JEO, KJY, LJFR. Resources: JJL, BK. Writing—original draft: DKE, JEO, KJY, LJFR, EJK, CRK, ALW. Writing—review and editing: DKE, JEO, KJY, LJFR, EJK, CRK, ALW, JJL, BK, JLSS. Visualisation: JEO, KJY, EJK. Supervision: EKJ, CRK. Project administration: JEO, EJK, CRK. Funding acquisition: LJFR.
Funding This work was supported by Exact Sciences.
Competing interests DKE is an employee of Exact Sciences Corporation. BK and JJL are employees of Exact Sciences Laboratories. LJFR offers scientific input to research studies through a contracted services agreement between Mayo Clinic and Exact Sciences. All other authors report no competing interests.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. Individual participant data that underlie the results reported in publications of the study will be shared after de-identification. This may include text, tables, figures and appendices. The study protocol (including statistical methods), informed consent form and clinical study report will also be shared. Data will be shared with researchers who provide a methodologically sound proposal to achieve the aims outlined in the approved proposal. Data will be available from 2 years and ending 4 years after publication. Proposals for access to data should be directed to clinicaltrials@exactsciences.com. To gain access, data requestors will need to provide a methodologically sound proposal and sign a data access agreement. Researchers are required to obtain necessary IRB/EC approvals or waivers as applicable to conduct research.