Article Text

Abstract
Introduction This randomised controlled trial aims to compare the efficacy of warfarin replacement with apixaban without postoperative resumption of heparin with that of conventional heparin bridging in patients taking warfarin and scheduled to undergo endoscopic mucosal resection (EMR): specifically, by checking for reduction in proportion of postoperative bleeding following EMR.
Methods and analysis The multicentre, open-label randomised parallel-group, controlled intervention study regards patients taking warfarin who are scheduled to undergo colon EMR. Enrolled patients have been taking oral warfarin for non-valvular atrial fibrillation or venous thrombosis on an outpatient basis, and have been found to have a colorectal polyp (including adenoma or early-stage cancer) for which EMR was indicated. They are assigned to one of two groups.
The primary endpoint is proportion of postoperative bleeding. The secondary endpoints are hospital stay length, therapeutic endoscopy outcomes such as proportion of en bloc resection, proportion of perforation, proportion of intraoperative bleeding, incidence of cerebral infarction/systemic embolism events, incidence of adverse events and serious adverse events, and proportion of postoperative therapeutic endoscopy.
Ethics and dissemination This trial was approved by the National Hospital Organization Central Review Board for Clinical Trials (19 April 2016).
Discussion The paucity of reports with high evidence levels was considered problematic when the Japan Gastroenterological Endoscopy Society’s gastroenterological endoscopy guidelines were revised in 2012. The results of this trial will have high medical significance, as its data could serve as the basis for revisions in the next edition of the guidelines.
Trial registration number This trial was registered in the University Hospital Medical Information Network Clinical Trials Registry (UMIN-CTR) accepted from the International Committee of Medical Journal Editors (ICMJE). UMIN000021947; Pre-results.
- endoscopic mucosal resection
- direct oral anticoagulant
- warfarin
- heparin
- apixaban
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
With the rapid ageing of Japanese society and increasing westernisation of dietary habits in recent years, more and more patients undergoing investigative or therapeutic endoscopies are being administered oral antithrombotics. Anticoagulants are the first choice for preventing cerebral infarction associated with atrial fibrillation, transient cerebral ischaemic attack, and other such cardiogenic cerebral embolisms. Warfarin and heparin have long been known to serve this purpose, but other, novel oral anticoagulants have been used since 2011. One of these is apixaban, the use of which has been on the rise due to its low risk of gastrointestinal bleeding and rapid onset of effects.1 In Japan Gastroenterological Endoscopy Society (JGES) guidelines published in 2012,2 endoscopic mucosal resection (EMR) is indicated for colon tumours based on high haemorrhagic risk of the procedure. Moreover, the guidelines deem heparin substitution followed by withdrawal,-that is , heparin bridging-to be necessary for patients taking oral anticoagulants. However, heparin bridging also faces problems: postoperative bleeding is reportedly significantly more common when it is used than when it is not,3 and it results in longer hospital stays. There are several reports on the risk of heparin bridging in Western countries.4 5 Birnie et al reported that heparin replacement did not decrease embolic events in patients who underwent insertion surgeries of pacemaker or implantable cardioverter defibrillator (ICD), while raising the incidence of device pocket haematoma.4 Moreover, Douketis et al reported that heparin replacement during a palliative surgery did not decrease thromboembolic events and increased the risk of major bleeding.5 However, there is no clinical trial that examined heparin bridging at EMR. Apixaban is considered to have a low risk of gastrointestinal bleeding among direct oral anticoagulants.6–9 In addition, since the maximum plasma concentration reaching time is 3–3.5 hours and the elimination half-life is 6–8 hours, it is unnecessary to administer heparin at the resumption and the risk of postoperative bleeding is considered to be low. Apixaban substitution is an alternative treatment for patients with atrial fibrillation taking oral warfarin at high risk of stroke: this perioperative procedure could result in lower proportion of postoperative bleeding and shorten hospital stay length following colon EMR.
Methods and analysis
Study objectives
This randomised controlled trial aims to compare the efficacy of warfarin replacement with apixaban without postoperative resumption of heparin with that of conventional heparin bridging in patients taking warfarin and scheduled to undergo EMR: specifically, by checking for reduction in proportion of postoperative bleeding following EMR.
Study design
This study is a multicentre, open-label, randomised, parallel-group, controlled intervention study (figure 1).

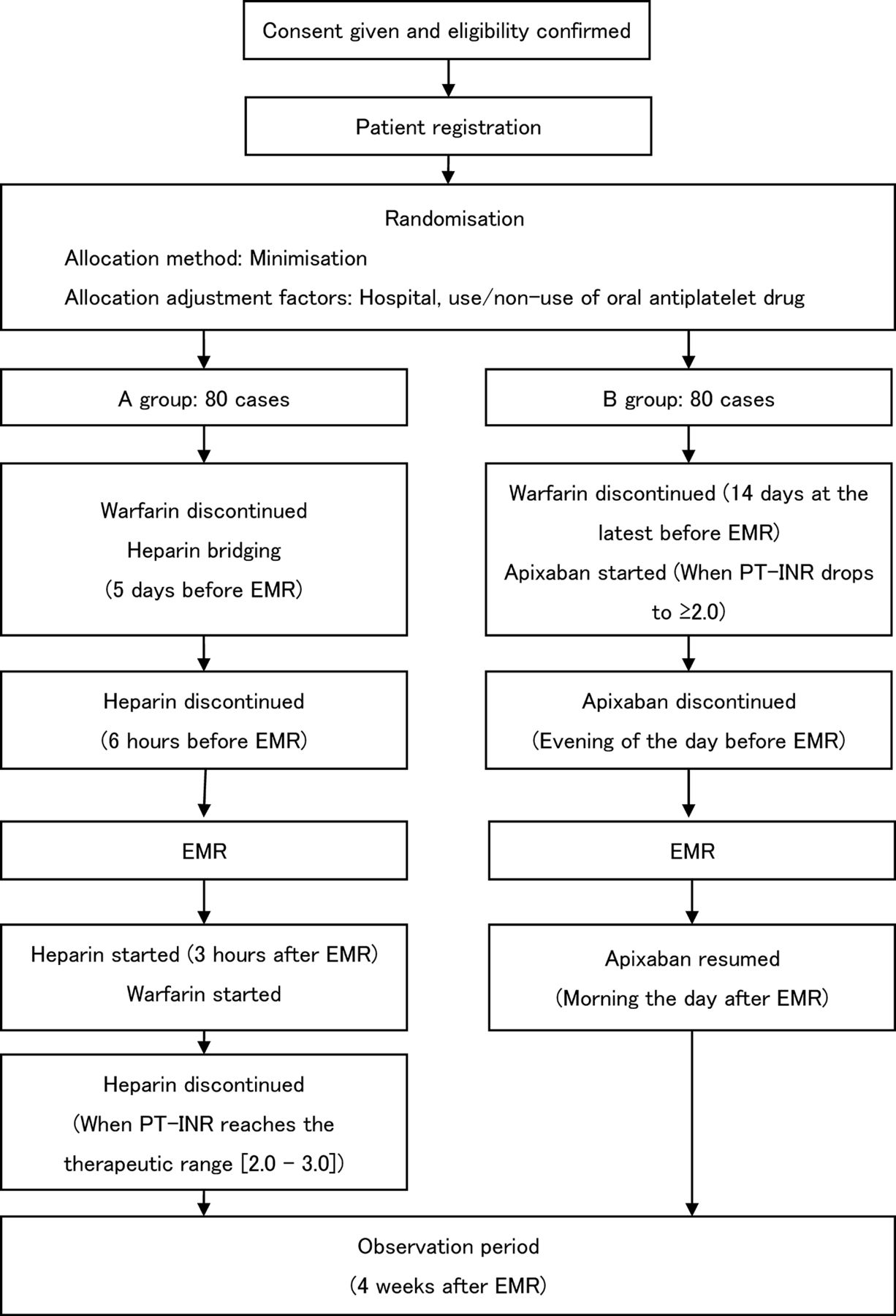{kind=link}
Entire design of this trail. EMR, endoscopic mucosal resection; PT-INR, prothrombin time-international normalised ratio.
Participating centres
National Hospital Organization Osaka National Hospital
National Hospital Organization Kyushu Medical Center
National Hospital Organization Kure Medical Center
National Hospital Organization Tokyo Medical Center
National Hospital Organization Mie Chuo Medical Center
National Hospital Organization Disaster Medical Center
National Hospital Organization Hakodate National Hospital
National Hospital Organization Hokkaido Cancer Center
National Hospital Organization Kanazawa Medical Center
National Hospital Organization Tsuruga Medical Center
National Hospital Organization Higashihiroshima Medical Center
Study population
Hypothesis is superiority of apixaban regimen group to warfarin group in reducing the proportion of postoperative bleeding. The study population consists of 160 patients, with 80 in standard treatment group (A group, ie, control group) and 80 in the experimental treatment group (B group).
Proportion of postoperative bleeding was supposed to be 20% in the standard treatment group, as prior studies have reported incidences of 10% to 23% after colon EMR with heparin bridging.4 10 It was supposed to be 5% in the experimental treatment group, based on the observation that none of the 10 patients who underwent EMR while taking direct oral anticoagulants at National Hospital Organization Osaka National Hospital from 2012 to 2014 experienced postoperative bleeding. Sample size was calculated as 76 per group given a one-sided significance level of 0.025 and statistical power of 0.80 to detect reduction in proportion of the experimental treatment group. In anticipation of dropouts, sample size has been set at n=80 (ie, 160 patients in total).
Subjects
Patients taking oral warfarin for non-valvular atrial fibrillation or venous thrombosis who have been found to have a colorectal polyp (including hyperplastic polyps, adenoma, or early-stage cancer) for which EMR was indicated.
Eligibility criteria
Inclusion criteria
Patients are included who fulfil all of the following criteria:
scheduled to undergo endoscopic mucosal resection for a colorectal polyp (including early-stage cancer)
taking oral warfarin for non-valvular atrial fibrillation or venous thrombosis, with prothrombin time-international normalised ratio (PT-INR) controlled at 3.0 or below
aged 20 years or older at the time of informed consent
provided written consent of his or her own free will.
Exclusion criteria
Patients are excluded who fulfil any of the following criteria:
having haemorrhagic diathesis (platelet count ≤5.0x104/L)
receiving maintenance dialysis
pregnant or potentially pregnant (women)
unable to reduce antiplatelet regimen to a single drug
deemed unsuitable for participation in this study by the principal investigator or other investigators.
Study outline
Treatment protocols
Patients taking oral warfarin for non-valvular atrial fibrillation or venous thrombosis on an outpatient basis who were found to have a colorectal polyp (including adenoma or early-stage cancer) for which EMR was indicated were assigned to one of the two groups below.
Standard treatment group (A group): Heparin bridging in patients receiving warfarin is performed according to the procedure in Guidelines for management of anticoagulant and antiplatelet therapy in cardiovascular disease. Patients are discontinued from warfarin and put on heparin on hospitalisation, 5 days before treatment. Any oral antiplatelet drugs being taken are suspended 1 day before treatment (3 days before if the drug is aspirin, and 5 days before if the drug is a thienopyridine derivative). However, patients who cannot discontinue their antiplatelet regimen are allowed to proceed in the cases of oral administration of aspirin and/or cilostazol. Regimens consisting of multiple oral antiplatelet drugs are replaced with an aspirin or cilostazol regimen. On the day of the operation, EMR is performed 6 hours after suspension of intravenous heparin administration. Heparin administration is resumed 3 hours after treatment. Patients resume oral warfarin at their preoperative dose the next day, once doctors confirm the absence of postoperative bleeding. Oral antiplatelet drugs discontinued prior to EMR are resumed at the same time as warfarin. PT-INR is monitored as appropriate; heparin administration is discontinued and the patient discharged once it reaches the therapeutic range (2.0–3.0). Each heparin regimen is begun at a dose of 10 000 units/day, and calibrated so that the patient’s activated partial thromboplastin time reaches 2.0 times its baseline value.
Experimental treatment group (B group): Patients are discontinued from warfarin 14 days before treatment, and started on apixaban once their PT-INR drops to ≥2.0. The standard apixaban dosage is 5.0 mg twice per day, but it is 2.5 mg twice per day for patients meeting at least two of these criteria: age ≥80 years, body weight ≤60 kg, or serum creatinine ≥1.5 mg/dL. Any oral antiplatelet drugs being taken are suspended 1 day before treatment (3 days before if the drug is aspirin, 5 days before if the drug is a thienopyridine derivative). However, patients who cannot discontinue their antiplatelet regimen are allowed to proceed in the cases of oral aspirin and/or cilostazol. Regimens consisting of multiple oral antiplatelet drugs are replaced with an aspirin or cilostazol regimen. Patients are hospitalised the day before treatment, and discontinued from apixaban that evening. EMR is performed the following day. Oral apixaban is resumed and the patient discharged once doctors confirm the absence of postoperative bleeding the morning after EMR. Oral antiplatelet drugs discontinued prior to EMR are resumed at the same time as apixaban.
Patients in both groups have an outpatient consultation 28 days after treatment to check for postoperative bleeding. In the event of positive findings, the haemorrhage site is verified using colonoscopy.
EMR was performed as follows: A sufficient volume of normal saline solution (with or without adrenalin) was injected through needle forceps into the submucosa under the polyp. The polyp was then grasped with a snare and excised by an electrosurgical generator. However, all were instructed to obey the following provisions:
no cold polypectomies
clipping after resection may be performed at the facility’s discretion.
Modified treatment schedule criteria
None specified.
Concurrent and supportive therapy
Colonoscopy is performed when hematochezia is confirmed, identifying the source of bleeding and performing haemostasis as much as possible.
Allocation
Patients are allocated to groups using an electronic data capture (EDC) system.
Method: Minimisation
Adjustment factors: Hospital, use/non-use of oral antiplatelet drugs
Data collection and monitoring
Data management
An EDC system is used in this study. Study investigators or those designated as such use strictly controlled personal electronic signatures (IDs and passwords) to log in to the EDC system. Collected patient information is promptly entered into the EDC and sent to a data centre. Transmitted electronic data are treated as case report forms. Study investigators or those designated as such perform all data entry and correction. They can make inquiries within the EDC system as necessary, as well as respond to inquiries created by data managers and other staff. Study investigators confirm the accuracy of all data input.
Monitoring
This trial employs centralised monitoring based on data collected via the EDC system; no on-site monitoring is performed in principle. Monitoring consists of checking data as necessary, and individual facilities may be contacted to add/enter any missing data. Periodic monitoring is done twice yearly.
Endpoints
Primary endpoint
Proportion of postoperative bleeding (postoperative bleeding is defined as overt hematochezia within the observation period (4 weeks) after EMR).
Secondary endpoints
hospitalisation period
therapeutic endoscopy outcomes (proportion of en bloc resection, proportion of perforation, proportion of intraoperative bleeding)
incidence of cerebral infarction/systemic embolism events
incidence of serious adverse events
proportion of postoperative therapeutic endoscopy.
Statistical analysis
Analysis sets
In accordance with the intention-to-treat (ITT) principle, the ITT set consists of all cases registered and allocated to groups in this study. The full analysis set (FAS) consists of all cases in the ITT set meeting the eligibility criteria for whom, at minimum, study treatment is started and at least one observation is recorded after randomisation. The safety analysis set consists of all cases who began the study treatment. Efficacy is primarily analysed in the FAS.
Primary endpoint analysis
Proportion of postoperative bleeding will be calculated for each group, and compared between groups by the χ2 test.
ORs for postoperative bleeding will be calculated using a logistic model including factors of group and whether or not oral antiplatelet drugs were administered.
Secondary endpoint analysis
Hospitalisation period will be compared between two groups by the Wilcoxon test.
For EMR outcomes, incidence of cerebral infarction/systemic embolism event, and proportion of postoperative therapeutic endoscopy, point estimates for differences of proportion and 95% CIs will be calculated.
Frequencies of adverse events and serious adverse events will be summarised in each group.
Interim analysis
No interim analysis will be performed.
Ethics and dissemination
Ethics
This study complies with the World Medical Association’s Declaration of Helsinki, Ethical Guidelines for Medical and Health Research Involving Human Subjects, and Act on the Protection of Personal Information. It was approved by the National Hospital Organization Central Review Board for Clinical Trials (19 April 2016).
Before participation in the study, patients must have the study fully explained to them by a study investigator or research team member using an explanation and consent form, and provide written consent of their own free will. The explanation and consent form meets the requirements of Ethical Guidelines for Medical and Health Research Involving Human Subjects, and its use has been approved by the Central Review Board for Clinical Trials.
Patient safety
Subjects will be monitored for adverse events by a study investigator or research team member. Wherever possible, they will record the diagnosis, not the individual signs and symptoms, in the case report if signs (including clinical test values) or symptoms are included in a diagnosis.
Serious adverse events will be treated immediately and appropriately: responding team members will make efforts to ensure the subject’s safety and investigate the event’s cause, and promptly report back to a study investigator.
Responses to and results of any serious adverse events will be promptly reported to the Minister of Health, Labour and Welfare, and announced on the hospital’s website if (1) the event was unpredictable and (2) researchers cannot rule out a causal relationship between the event and this study.
Discussion
Increasing numbers of patients are receiving anticoagulant therapy as society ages, putting more and more of them at haemorrhage risk. Gastrointestinal bleeding is rare in such patients who undergo therapeutic endoscopy. Doctors who suspend a patient’s anticoagulant therapy due to excess concern over gastrointestinal bleeding instead increase their risk of cerebral infarction, embolism, and other such serious events. Thus, both suspension and continuation of anticoagulant therapy can cause serious illness, leading to increases in hospitalisation and treatment costs. Heparin bridging is currently considered the standard treatment prior to therapeutic endoscopy for patients taking oral warfarin; however, higher proportion of postoperative bleeding and longer hospital stays associated with heparin bridging have also brought increases in hospitalisation costs and treatment costs. There are several reports on the risk of heparin bridging.4 5 However, since there is no clinical trial that examined heparin bridging at EMR, we believe that this trial is important. This study seeks to establish a procedure that presents lower risks of haemorrhage, thrombosis, and embolism, and which could lower medical costs by reducing the length of hospital stays. Assessing the effects and risks of apixaban’s use in therapeutic endoscopy for patients receiving anticoagulant therapies could also lead to safer and more effective ways of managing anticoagulant therapies. The paucity of reports with high evidence levels was considered problematic when the JGES gastroenterological endoscopy guidelines were revised in 2012. The results of this trial will have clinical significance, as its data could serve as the basis for revisions in the next edition of the guidelines.
References
Footnotes
Contributors TY participated in the design of the study. AK participated in the design and statistical analysis plan of the study. TY and AS are responsible for registering cases, managing data and handling central monitoring. TY, TK, TU, NW, YS, KM, YT, TK, TK, HH and NH conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Funding This study is being conducted using an operating expense grant for research from the National Hospital Organization.
Competing interests None declared.
Ethics approval This trial was approved by the National Hospital Organization Central Review Board for Clinical Trials (19 April 2016).
Provenance and peer review Not commissioned; externally peer reviewed.