Article Text

Statistics from Altmetric.com
- HCV, hepatitis C virus
- IR, insulin resistance
- NASH,
- non-alcoholic steatohepatitis,
- MTP, microsomal transfer protein
- VLDL, very-low density lipoproteins
- SCD4, stearoyl coenzyme A desaturase 4
- ob (obese),
- BMI, body mass index
- IRS, insulin receptor substrate
- SREBP-1, sterol regulatory element binding protein-1c
- SOC, suppressor of cytokine signaling
- PI3K, phosphatidylinositol 3-kinase
- STAT-3, signal transducer and activator of transcription-3
- TNF, Tumor necrosis factor
- PEPCK, phosphoenolpyruvate carboxykinase
The relationship between hepatitis C virus (HCV), steatosis, and insulin resistance is genotype specific, and steatosis and insulin resistance are closely linked to the progression of liver disease in HCV infected patients
Since the identification of hepatitis C virus (HCV) in the late 1980s, chronic HCV infection has emerged as a complex multifaceted disease with manifestations extending beyond the liver. As such, hepatic steatosis, insulin resistance (IR), and type II diabetes have been observed to occur more frequently in association with HCV infection than other chronic inflammatory liver disease.1 A proportion of HCV infected patients with steatosis also exhibit several of the clinical features seen in non-alcoholic steatohepatitis (NASH), questioning the significance of these metabolic disorders in the pathogenesis of HCV related liver disease. Hence, considerable HCV research has recently been directed towards understanding the mechanisms underlying the development of these metabolic manifestations in HCV infected patients and their implications in the progression of liver disease. Several important questions have been examined: are these metabolic disorders in HCV infected patients a result of viral or host factors and, if viral, how do viral proteins interfere with lipid and insulin metabolism? What is the primary event in these patients (steatosis or IR) and what are the implications of steatosis and IR in the pathogenesis of liver disease? Finally, how can we exploit our current knowledge for developing effective therapeutic strategies for HCV infected patients?
In this issue of Gut, Fartoux and colleagues2 utilise the homeostasis model assessment for IR (HOMA IR) index to study the association between steatosis and IR in patients with chronic HCV infection (see page 1003). In order to examine this association, the authors excluded patients with alternate factors known to be associated with the development of steatosis (such as alcohol intake >20 g/day or diabetes). Their findings confirm earlier observations3–5 that differing and genotype specific mechanisms characterise the development of hepatic steatosis in HCV infected patients. Accordingly, steatosis in genotype 3 infected patients is primarily viral mediated (cytopathic) whereas host factors, principally those associated with IR and its clinical attributes, are responsible for the development of steatosis in the genotype 1 infected patient.
DIRECT STEATOGENIC EFFECT OF HCV
Evidence for a HCV genotype 3 specific steatogenic effect has come from several clinical observations (table 1).3,6–8 Furthermore, this direct steatogenic effect of HCV is highly reproducible in transgenic mice and cell culture models of hepatitis C, in which overexpression of viral protein (such as core and NS5A) has been shown to induce accumulation of intracytoplasmic triglyceride rich droplets.9
Clinical evidence for a steatogenic effect for genotype 3
There are several identified mechanisms whereby HCV may alter lipid metabolism. Firstly, HCV core protein has been shown to directly inhibit the function of microsomal triglyceride transfer protein, a major regulator of hepatic assembly, and secretion of nascent triglyceride rich very low density lipoproteins (VLDL). The latter effect impairs the ability of hepatocytes to assemble and secret VLDL.10,11 Secondly, HCV core protein has been observed to induce mitochondrial injury resulting in oxidative stress. Oxidative stress perturbs lipid peroxidation, thereby contributing to the development of steatosis.12–14 Finally, microarray studies have illustrated the ability of HCV (in particular genotype 3) to induce transcription of several genes involved in lipid metabolism in the liver.15,16 Among these is stearoyl coenzyme A desaturase 4 (SCD4), a rate limiting enzyme in the synthesis of monounsaturated fats.15 Reduced expression of SCD4 in livers of ob/ob mice has been shown to significantly ameliorate hepatic steatosis.17
INSULIN RESISTANCE AND STEATOSIS IN HCV INFECTED PATIENTS
Steatosis in patients with genotype 1 infection is increasingly recognised as a component of the metabolic syndrome, a condition invariably associated with IR. In support, host factors such as age, body mass index (BMI), and central obesity, have been shown to correlate with the development of steatosis in genotype 1 infected patients (but not genotype 3).18 Fartoux and colleagues2 have further demonstrated that HOMA IR was the only risk factor independently associated with the development of steatosis in genotype 1 infected patients (p = 0.001). Moreover, the degree of steatosis was significantly predictive of HOMA IR (p = 0.004).
There are several uncertainties regarding the cascade of events leading to the development of IR and steatosis in HCV infected patients with features of the metabolic syndrome. As such, whether IR or steatosis is the primary or secondary event in these patients is unclear.
Overall, there are sufficient clinical and experimental data indicating that excess fat can perpetuate IR: large epidemiological studies reveal that the risk of IR rises as body fat content (determined by BMI) increases, from the very lean to the very obese.19 Experimental evidence also indicates that intracellular accumulation of fatty acid metabolites (either through increased delivery or decreased metabolism) in the liver or muscle directly promotes serine phosphorylation (in contrast with tyrosine phosphorylation) of insulin receptor substrate 1 (IRS-1). This effect leads to impaired glucose transport activity and other events downstream of insulin receptor signalling.20 In this respect, measures to reduce circulating free fatty acid and liver triglyceride content have been shown to restore insulin signalling and reverse both hepatic and muscle IR induced by high fat diets.17,21
Another common link in the development of IR and steatosis is the adipocyte secreted proteins leptin and adiponectin. Leptin is a protein product of the adipocyte obese (ob) gene. Serum leptin levels have been observed to be increased in patients with NASH and chronic HCV infection.22,23 Moreover, in each of these diseases, serum leptin level has been reported to independently correlate with hepatic steatosis.22,24 While it is recognised that the effect of leptin on insulin sensitivity is variable, increased expression of leptin in hepatocytes has been shown to attenuate several insulin induced activities, including tyrosine phosphorylation of IRS-1.25
Adiponectin is abundantly synthesised and secreted by adipose tissue. Serum levels of adiponectin correlate with systemic insulin sensitivity. More recently, low levels of serum adiponectin have been shown to correlate with the presence of steatosis in patients with chronic HCV infection. The latter observation suggests a role for adiponectin in the development of IR and steatosis in these patients.26
In contrast with the effect of fat on IR, insulin itself controls the regulation of a host of genes involved in lipid metabolism.27 The ability of insulin to activate lipogenesis is partly mediated by inducing the transcription of the sterol regulatory element binding protein 1c (SREBP-1c), a key regulator of fatty acid synthesis in the liver. The effect of insulin on SREBP-1c transcription is even more apparent in the presence of profound IR.28 In this setting, obese animals with IR have increased levels of SREBP-1c resulting in increased rates of fatty acid synthesis and the development of hepatic steatosis.29,30 Subsequent normalisation of SREBP-1 expression in these animals dramatically ameliorates steatosis.28
HCV AND INSULIN RESISTANCE
Several studies (including that of Fartoux and colleagues2) evaluating IR in patients with chronic HCV infection have found that the development of IR can occur early in the course of the disease. This effect appears to be independent of body weight, stage of liver disease, and presence or absence of overt diabetes.31–33 There are other clinical observations supporting a “fat independent” mechanism in the development of IR in HCV infected patients. Firstly, Fartoux and colleagues2 and others32 have observed that patients infected with HCV genotype 3 have more extensive hepatic steatosis but a lower incidence of IR.32 This observation supports a genotype specific mechanism underlying IR in HCV infected patients.32 Secondly, two studies demonstrated that clearance of HCV with antiviral therapy resulted in restoration of insulin sensitivity and euglycaemia.34,35 Finally, the extent of IR has been shown to correlate with the grade of portal inflammation in HCV infected patients.32 Collectively, these data suggest that either HCV per se or the inflammatory response incited by HCV in the liver is central to the development of IR. This first hypothesis has been recently verified in HCV core transgenic mice, observed to develop IR resistance as early as one month of age, in the absence of either overt liver injury or excessive body weight gain.
The precise mechanisms whereby HCV induces IR remain elusive but recent progress has shed light on several critical pathways. Impairment of IRS-1 and IRS-2 expression has been observed in the livers of both HCV infected patients as well as in HCV core transgenic mice.36,37 Specifically, HCV core protein has been shown to inhibit insulin induced phosphorylation of the p85 subunit of phosphatidylinositol 3-kinase (PI3K) and Akt, which are downstream components of IRS in the liver.37 Interestingly, HCV has been reported to mediate dysfunction of the insulin signalling pathways by upregulating the expression of suppressor of cytokine signalling 3 (SOC3).37 This observation is quite important in light of recent data demonstrating direct involvement of SOC1 and SOC3 in regulating expression of SREBP-1c in the livers of obese animals.38 In these animal models, overexpression of SOC1 and SOC3 proteins has been shown to enhance SREBP-1c promoter activity by attenuating signal transducer and activator of transcription 3 (STAT-3) mediated inhibition of this region.38 The net effect in these animals was development of systemic IR and hepatic steatosis. Conversely, inhibiting expression of SOCs protein in obese animals normalised levels of SREBP-1 and improved insulin sensitivity and hepatic steatosis. Consistent with these data, hepatic STAT-3 signalling has recently been shown to be essential for normal glucose homeostasis and insulin sensitivity.39 These observations are relevant as HCV is recognised as influencing the activity of STAT-3. It is therefore intriguing to speculate that STAT-3 may be one of the key molecules involved in HCV mediated IR (fig 1).

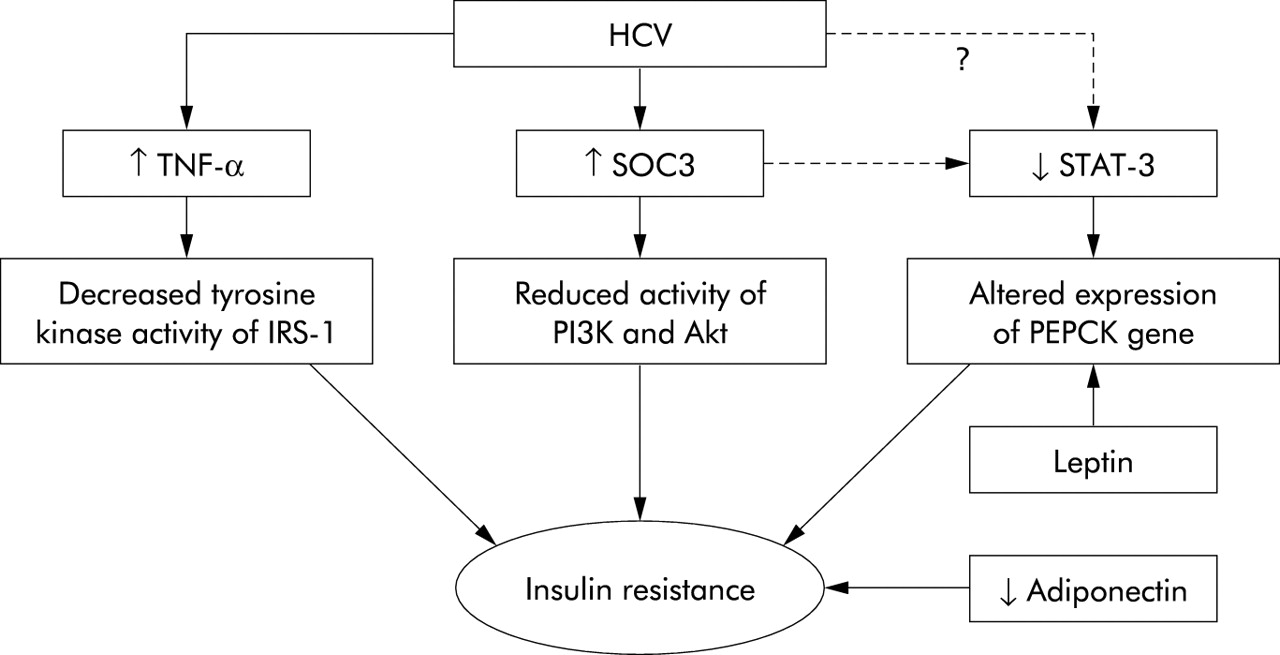{kind=link}
Molecules that are likely to be involved in mediating insulin resistance (IR) in hepatitis C virus (HCV) infection. Tumour necrosis factor α (TNF-α) is increased in the serum and liver of HCV infected patients. TNF induces IR by decreasing the tyrosine kinase activity of insulin receptor substrate 1 (IRS-1).39 Treatment of HCV core transgenic mice with anti-TNF-α has been shown to restore insulin sensitivity. High levels of suppressor of cytokine signalling 3 (SOC3) have been detected in association with IR in HCV infection. This effect was associated with reduced insulin induced phosphorylation of the p85 subunit of phosphatidylinositol 3-kinase (PI3K) and Akt.37 SOC3 (and possibly HCV) can attenuate the activity of signal transducer and activator of transcription 3 (STAT-3). Mice lacking STAT-3 specifically in the liver have been reported to exhibit IR and, when fed a high fat diet, glucose intolerance. The latter was associated with increased expression of the phosphoenolpyruvate carboxykinase (PEPCK) gene.38 The PEPCK gene itself is a target for leptin (also increased in HCV infection) mediated regulation of gluconeogenesis in the liver. Finally, reduced expression of adiponectin is associated with obesity and IR. Low serum levels of adiponectin have been shown to correlate with steatosis in HCV infected patients.26
ROLE OF CYTOKINES
Tumour necrosis factor α (TNF-α) levels are elevated in the liver and serum of patients with chronic HCV infection. TNF-α plays an important role in the development of IR through interfering with insulin signalling.40 In IR HCV core transgenic mice, treatment with anti-TNF-α restored insulin sensitivity, thus supporting the notion that TNF-α is a major factor for the development of IR in HCV infection.31
ROLE OF STEATOSIS AND INSULIN RESISTANCE IN PROGRESSION OF HCV LIVER DISEASE
In agreement with previous reports,3,4,7,41 Fartoux and colleagues2 observed an association between steatosis and severity of fibrosis, irrespective of HCV genotype. While insulin levels were predictive of fibrosis in their univariate analysis, subsequent multivariate analysis confirmed steatosis, but not insulin levels, to be independently associated with fibrosis (p = 0.02).
Thus two keys questions arise: is fat per se fibrogenic and what is the role of insulin in the fibrotic process?
Steatosis is associated with increased production of reactive oxygen species which initiate lipid peroxidation, resulting in hepatic stellate cell activation.42 However, in NASH, disease progression is recognised as being slower than that observed in patients with HCV infection and steatosis. Thus it is likely that the coexistence of HCV and steatosis aggravates and accelerates the injury induced by each alone. In this setting, hepatic inflammation induced by the host response, together with the increased production of several proinflammatory and profibrotic cytokines, provide the substrate for the “second hit” in the steatotic liver. Also, the ability of the virus itself to induce oxidative stress and promote lipid peroxidation may further aggravate the pathogenic process induced by steatosis. Consider also that fat may render HCV infected liver more vulnerable to injury. Livers with steatosis are more sensitive to TNF-α mediated inflammation and liver injury.43 Moreover, in HCV livers with steatosis, apoptosis activity has been noted to be increased compared with infected livers without steatosis.44
While Fartoux and colleagues2 could not find a direct association between IR and fibrosis, others have.32,33,45 The authors do concede that insulin plays a significant role in the development of fibrosis via a mechanism involving steatosis. In this regard, steatosis promotes cellular IR which, in turn, induces compensatory hyperinsulinaemia. Hyperinsulinaemia has been shown to directly stimulate hepatic stellate cell proliferation and increase expression of connective tissue growth factor, a key factor in the progression of fibrosis.46 Collectively, the data implicate both steatosis and IR in liver disease progression in HCV infected patients.
CONCLUSIONS
Our knowledge of the mechanisms and significance of steatosis and IR in patients with chronic HCV infection has advanced. The work by Fartoux and colleagues2 adds to the growing evidence that the relationship between HCV, steatosis, and IR is genotype specific and that steatosis and IR are closely linked to the progression of liver disease in HCV infected patients. As such, we need to clarify the molecular pathways involved in mediating these inter-relationships. In light of our current knowledge of the implications of steatosis and IR in liver disease, the importance of lifestyle changes (such as weight loss) need to be an emphasised in treating HCV infected patients. Similarly, new therapeutic approaches exploiting the interaction between HCV and lipid metabolism are eagerly awaited.
Acknowledgments
This work was supported by a grant from NIH, 2RO1 DK053792.
The relationship between hepatitis C virus (HCV), steatosis, and insulin resistance is genotype specific, and steatosis and insulin resistance are closely linked to the progression of liver disease in HCV infected patients
REFERENCES
Footnotes
-
Conflict of interest: None declared.