Article Text

Abstract
Objective Serrated polyps (hyperplastic polyps, sessile or traditional serrated adenomas), which can arise in a sporadic or polyposis setting, predispose to colorectal cancer (CRC), especially those with microsatellite instability (MSI) due to MLH1 promoter methylation (MLH1me+). We investigate genetic alterations in the serrated polyposis pathway.
Design We used a combination of exome sequencing and target gene Sanger sequencing to study serrated polyposis families, sporadic serrated polyps and CRCs, with validation by analysis of The Cancer Genome Atlas (TCGA) cohort, followed by organoid-based functional studies.
Results In one out of four serrated polyposis families, we identified a germline RNF43 mutation that displayed autosomal dominant cosegregation with the serrated polyposis phenotype, along with second-hit inactivation through loss of heterozygosity or somatic mutations in all serrated polyps (16), adenomas (5) and cancer (1) examined, as well as coincidental BRAF mutation in 62.5% of the serrated polyps. Concurrently, somatic RNF43 mutations were identified in 34% of sporadic sessile/traditional serrated adenomas, but 0% of hyperplastic polyps (p=0.013). Lastly, in MSI CRCs, we found significantly more frequent RNF43 mutations in the MLH1me+ (85%) versus MLH1me− (33.3%) group (p<0.001). These findings were validated in the TCGA MSI CRCs (p=0.005), which further delineated a significant differential involvement of three Wnt pathway genes between these two groups (RNF43 in MLH1me+; APC and CTNNB1 in MLH1me−); and identified significant co-occurrence of BRAF and RNF43 mutations in the MSI (p<0.001), microsatellite stable (MSS) (p=0.002) and MLH1me+ MSI CRCs (p=0.042). Functionally, organoid culture of serrated adenoma or mouse colon with CRISPR-induced RNF43 mutations had reduced dependency on R-spondin1.
Conclusions These results illustrate the importance of RNF43, along with BRAF mutation in the serrated neoplasia pathway (both the sporadic and familial forms), inform genetic diagnosis protocol and raise therapeutic opportunities through Wnt inhibition in different stages of evolution of serrated polyps.
- CANCER GENETICS
- CANCER SYNDROMES
- COLONIC POLYPS
- COLORECTAL CANCER GENES
- GENETIC TESTING
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
Serrated polyps can arise in a sporadic or familial polyposis setting and predispose to colorectal cancer (CRC), especially those with microsatellite instability (MSI) due to MLH1 promoter methylation (MLH1me+), whereas germline DNA mismatch repair gene mutations cause MSI CRC without MLH1 promoter methylation.
A recent study reported observing frequent somatic RNF43 frameshift mutation in coding mononucleotide repeats in CRC with microsatellite instability that was mutually exclusive with APC mutation, whereas frequent RNF43 mutation was recently reported in traditional serrated adenomas.
Germline RNF43 mutation has been recently reported in four individuals from three families that manifest serrated polyposis, but further study is needed to confirm its pathogenicity.
RNF43 is an E3 ubiquitin ligase that acts as a Wnt inhibitor by targeting Wnt receptors for degradation.
What are the new findings?
Second-hit inactivation of RNF43 was observed in all serrated polyps, adenomas and cancers arising from germline RNF43 mutation carriers, the latter cosegregates with the serrated polyposis phenotype.
RNF43 is mutated somatically with predominant two-hit inactivation in 34% of sessile serrated adenomas (SSAs)/traditional serrated adenomas, but not in hyperplastic polyps, whereas an SSA with CpG island methylator phenotype and focal MLH1 protein loss shows early emergence of RNF43 mononucleotide repeat tract mutation.
In MSI CRC, RNF43 mutation is more frequent in the MLH1me+ (85%) versus MLH1me− (33.3%) group, whereas APC/β-catenin mutation is more frequent in the latter. There is a strong tendency for co-occurrence of RNF43 with BRAF mutation in MSI and microsatellite stable (MSS) CRCs.
RNF43 mutation in human SSA was shown to have reduced dependency on R-spondin.
How might it impact on clinical practice in the foreseeable future?
Our results expand on previous studies to provide unequivocal proof of the pathogenicity of RNF43 germline mutation, establishing the need for routine germline testing for RNF43 mutation in serrated polyposis syndrome.
RNF43 mutation serves as a biomarker for progression from sporadic hyperplastic polyps to sessile/traditional serrated adenomas.
Our study has identified a distinct role for RNF43 in the serrated neoplasia pathway. Given that previous cell line and animal models have shown responsiveness of RNF43 mutant tumours to pharmacological Wnt secretion inhibitors in the preclinical or clinical phase of development, our findings point towards therapeutic opportunities through Wnt inhibition in different stages of evolution of serrated polyps.
Our comprehensive delineation of the differential involvement of APC/β-catenin/RNF43 and corresponding KRAS/BRAF alterations in different pathways of colorectal carcinogenesis provide a roadmap to guide therapeutic development targeting these pathways.
Introduction
Colorectal cancer (CRC) progresses through two major pathways, the adenoma-carcinoma sequence or through serrated polyps. Most microsatellite stable (MSS) CRCs occur due to APC gene mutation as an early event leading to aberrant Wnt activation. Alternatively, sporadic CRCs with microsatellite instability (MSI) may arise from serrated polyps, spanning a morphological spectrum from hyperplastic polyps (HPs), to sessile serrated adenomas (SSAs) and traditional serrated adenomas (TSAs), with SSAs/TSAs representing more advanced lesions with higher propensity for malignant progression.1 ,2 Previously, we discovered more frequent BRAF mutations in the progression from HPs to serrated adenomas,3 which has subsequently become the molecular marker for sporadic CRCs progressing through the serrated neoplasia pathway. These serrated polyps can then develop into MSI CRCs through acquisition of CpG island methylator phenotype (CIMP) and methylation of the MLH1 promoter, or MSS CRCs. Conversely, patients with Lynch syndrome with germline DNA mismatch repair (MMR) gene mutation develop CRC with MSI through the adenoma-carcinoma sequence.4 However, a substantially lower incidence of APC mutation in MSI CRCs5 ,6 casts doubt on whether Wnt activation is necessary for this group.
While serrated polyps are common and mostly sporadic in nature, patients with serrated (hyperplastic) polyposis syndrome are characterised by numerous (>20) or large proximal serrated polyps in the colon, and a smaller number of tubular or villous adenomas.2 ,7 The prevalence has been estimated to be as high as 1 in 3000 in some populations. Individuals and first-degree relatives have an increased risk of developing a variable number of serrated polyps or CRC, predominantly of MSS8 ,9 but occasionally of MSI types,10 but the genetic basis or mode of inheritance remains unclear. Recently, a whole-exome sequencing study in a series of patients with serrated polyposis identified mutations in several putative genes (ATM, PIF1, TELO2, XAF1, RBL1 and RNF43) functionally related to oncogenic induced senescence.11 Interestingly, two unrelated patients shared the same germline nonsense mutation in RNF43. Another study reported a family with two siblings carrying germline nonsense RNF43 mutation and numerous serrated polyps at a young age, one of whom developed a MSS CRC.12 However, the role of the germline RNF43 mutation needs further validation, as these families were small and cosegregation information was insufficient. RNF43 is an E3 ubiquitin ligase expressed in colon stem cells that acts as a Wnt inhibitor by targeting Wnt receptors for degradation. It regulates Wnt signal strength through the R-spondin/LGR5/RNF43 module, with its effect antagonised by the Wnt amplifier R-spondin.13–15 Recent studies identified frequent RNF43 mutation in MSI colorectal, gastric and endometrial cancers, with mutational hot spots residing in two short mononucleotide repeats which are likely a consequence of MSI.16 ,17 MSI CRCs constitute two distinct groups, those with MLH1 promoter methylation (a hallmark of the serrated neoplasia pathway) and those without (a consequence of germline or rarely somatic MMR gene mutation).18 ,19 BRAF mutation is associated specifically with the methylated group,20 but other differences between these two groups in terms of cancer driver genes or pathway mutations is unknown. Discovery of Wnt upstream regulators in these various types of lesions present therapeutic opportunities as clinical trials of Wnt secretion inhibitors is ongoing.
Methods
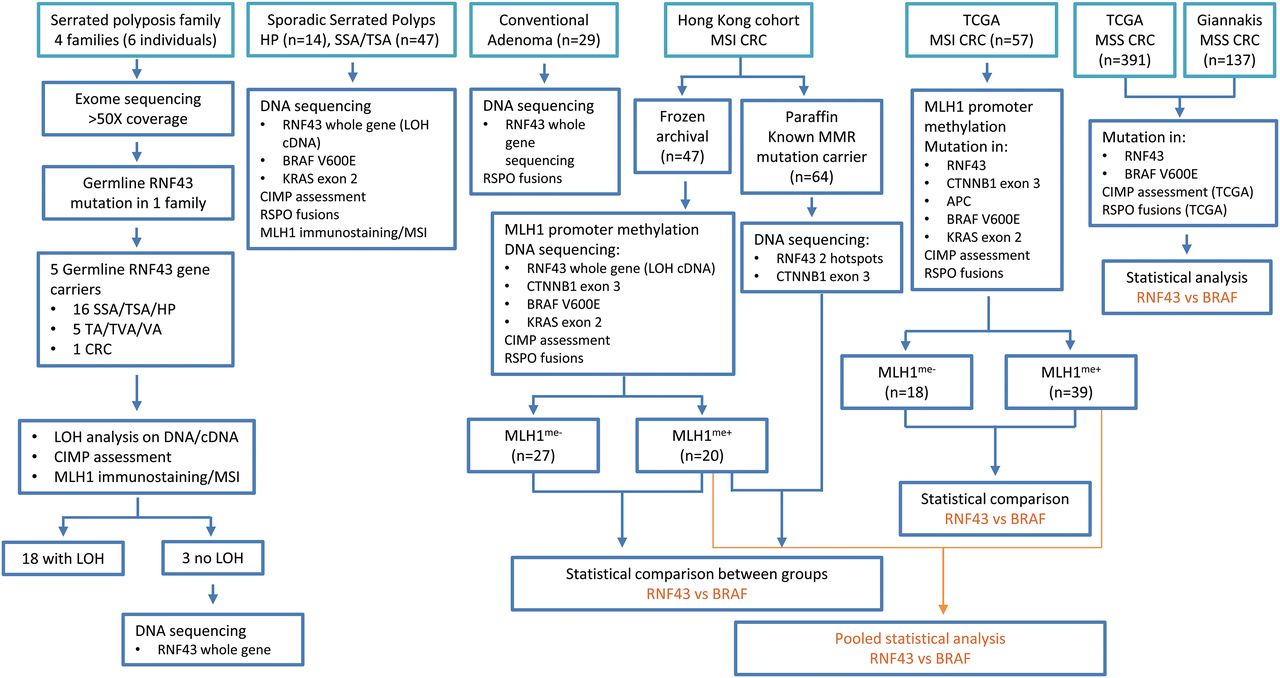
Figure 1 summarises the different study cohorts and their corresponding analysis flow chart.
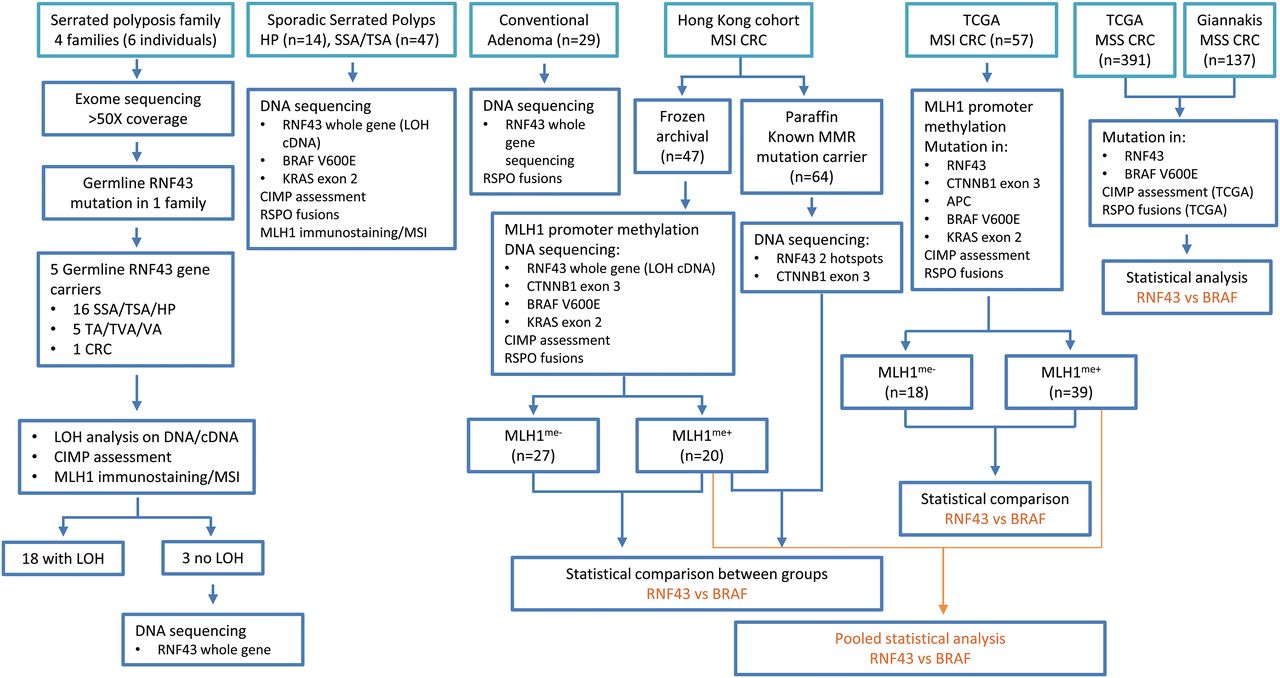
Summary of different patient cohorts, serrated polyps, conventional adenomas and colorectal cancer samples, and their corresponding analysis flow chart. CIMP, CpG island methylator phenotype; CRC, colorectal cancer; LOH, loss of heterozygosity; MSI, microsatellite instability; MSS, microsatellite stable; SSA, sessile serrated adenoma; TA, tubular adenoma; TSA, traditional serrated adenoma; TVA, tubulovillous adenoma; VA, villous adenoma.
See online supplementary methods for details on whole-exome sequencing, RNF43 somatic mutational analysis, mutational analysis of BRAF, KRAS, CTNNB1 hot spots, MLH1 promoter methylation analysis by pyrosequencing, assessment for CIMP, R-spondin (RSPO) fusions, MSI analysis, MLH1 immunostaining, bioinformatics analysis on The Cancer Genome Atlas (TCGA) cohort including somatic variant calling, fusion transcript detection, analysis of methylation arrays, establishment of organoid culture from patient, creation of ZNRF3−/RNF43− organoids in mice and cell viability assay.
Supplementary methods
Results
Exome sequencing identifies germline mutation of RNF43 in a family that cosegregates with a serrated polyposis phenotype in an autosomal dominant pattern
We examined a series of patients with serrated polyposis by whole-exome sequencing (six patients from four families, see online supplementary table S1 for clinicopathological data, figure 1 for analysis flow chart) with a mean coverage above 50× (see online supplementary table S2). We identified 274 germline variants with putative deleterious impact to gene function based on a combination of stringent bioinformatic algorithms (see online supplementary methods, online supplementary table S3). In one family, we found a germline RNF43 mutation (c.953-1, G>A) involving an essential splice site which was confirmed to cause aberrant splicing and a 2 bp deletion in the RNA (c.953_954delAG), thereby resulting in a truncated protein product (E318fs) (see online supplementary figure S1). The mutation was detected in three siblings in the family, two with serrated polyposis (patient A:II-2, A:II-3), the third with rectal cancer at age 49 years and a HP in the descending colon (A:II-6) (figure 2A and see online supplementary table S1). Starting at age 65 years, patient A:II-2 had numerous polyps (>100) detected in his colon, predominantly SSAs, some with transitional morphology to TSAs, HPs, as well as a smaller number of tubular or villous adenomas. Patient A:II-3 also had more than 20 SSAs or HPs distributed all over the colon. Subsequent colonoscopy examination of two children of A:II-6, both confirmed gene carriers, showed several SSAs or HPs at a young age (ages 37 years and 35 years), with some located proximal to the sigmoid colon. These five confirmed gene carriers all satisfied the WHO criteria of serrated polyposis. One gene carrier (A:II-5, age 60 years) had a comorbid condition which precluded colonoscopy, even though a clinical history of colonoscopy performed at age 44 years reported a normal finding. The other four family members that did not carry the germline mutation had normal colonoscopies from ages 41 years to 56 years.
Supplementary tables
Supplementary figures

(A) Pedigree of a serrated polyposis family (family A) with germline RNF43 mutation. Number in bracket, age of onset in years; SP, serrated polyposis phenotype satisfying WHO 2010 criteria; PA, pituitary adenoma; CRC, colorectal cancer; +/−, germline RNF43 mutation status; individual II-7, II-8, II-9 and III-3 had normal colonoscopies at age 56 years, 52 years, 46 years and 41 years, respectively. II-5 had a history of normal colonoscopy at age 44 years, but a colonoscopy could not be repeated (current age 60 years) because of a comorbid condition. (B) Second somatic hit in polyps or cancer from representative family members, their corresponding histology, sequence chromatogram and mutation status of BRAF or KRAS. SSA, sessile serrated adenoma; TA, tubular adenoma; TSA, traditional serrated adenoma; TVA, tubulovillous adenoma; VA, villous adenoma; LOH, loss of heterozygosity. Scale bar represents 500 µm. (C) Summary of incidence of RNF43 2nd hits, BRAF and KRAS mutations, and CpG island methylator phenotype (CIMP) status in 22 colorectal polyps/cancers from family members separated into pathological categories. See online supplementary table S4 for details.
RNF43 second hit by loss of heterozygosity or somatic mutation observed in all serrated polyps, adenomas and cancer in germline mutation carriers
To confirm the biological significance of the germline mutation, a total of 16 serrated polyps (SSAs, TSAs or HPs), 5 adenomas (tubular, tubulovillous or villous) and 1 cancer from these five gene carriers were characterised in detail for RNF43 somatic inactivation events, BRAF and KRAS mutations. All showed second-hit inactivation, either by loss of heterozygosity in DNA and/or RNA, or second somatic RNF43 mutations (figure 2), including a nonsense mutation (c.337C>T, R113X), a frameshift mutation (c.1322_23 insC, P441fs) or a missense mutation with high functional significance (c.461 C>T, P154L), with the latter observed in two separate SSAs (figures 2 and 3A, see online supplementary figure S2, online supplementary table S4). These data support unequivocally that RNF43 second hit contributes significantly to serrated polyps, adenomas or cancer development, and therefore the pathogenic role of the germline mutation. Of the serrated polyps 62.5% carried BRAF V600E mutation, another 18.75% carried KRAS mutation. The five adenomas did not carry BRAF mutation, but a villous adenoma had a KRAS mutation. The rectal cancer was wild type for BRAF or KRAS. All serrated polyps, adenomas and cancers were MSS and showed intact MLH1 protein expression. Eight out of 13 serrated polyps examined (61.5%) showed high level CIMP (hereby referred to as CIMP-positive), whereas the conventional adenomas and CRC did not display CIMP (figure 2C). Our data indicate that while RNF43 inactivation can give rise to both serrated polyps and tubular or villous adenomas, there is a preponderance towards development of serrated polyps with coincidental BRAF mutation. Interestingly, some of the serrated polyps in patients A:II-2 and A:II-3 showed increased number of Paneth cells (see online supplementary figures S2G and S3), mimicking the morphology of adenomas developed in RNF43/ZNRF3 double-knockout mice, even though we did not detect ZNRF3 mutation in these patients.15 Paneth cells were also noted in morphologically normal colonic crypts adjacent to the serrated polyps, raising the interesting possibility that these may provide the source of Wnt to drive cell proliferation.
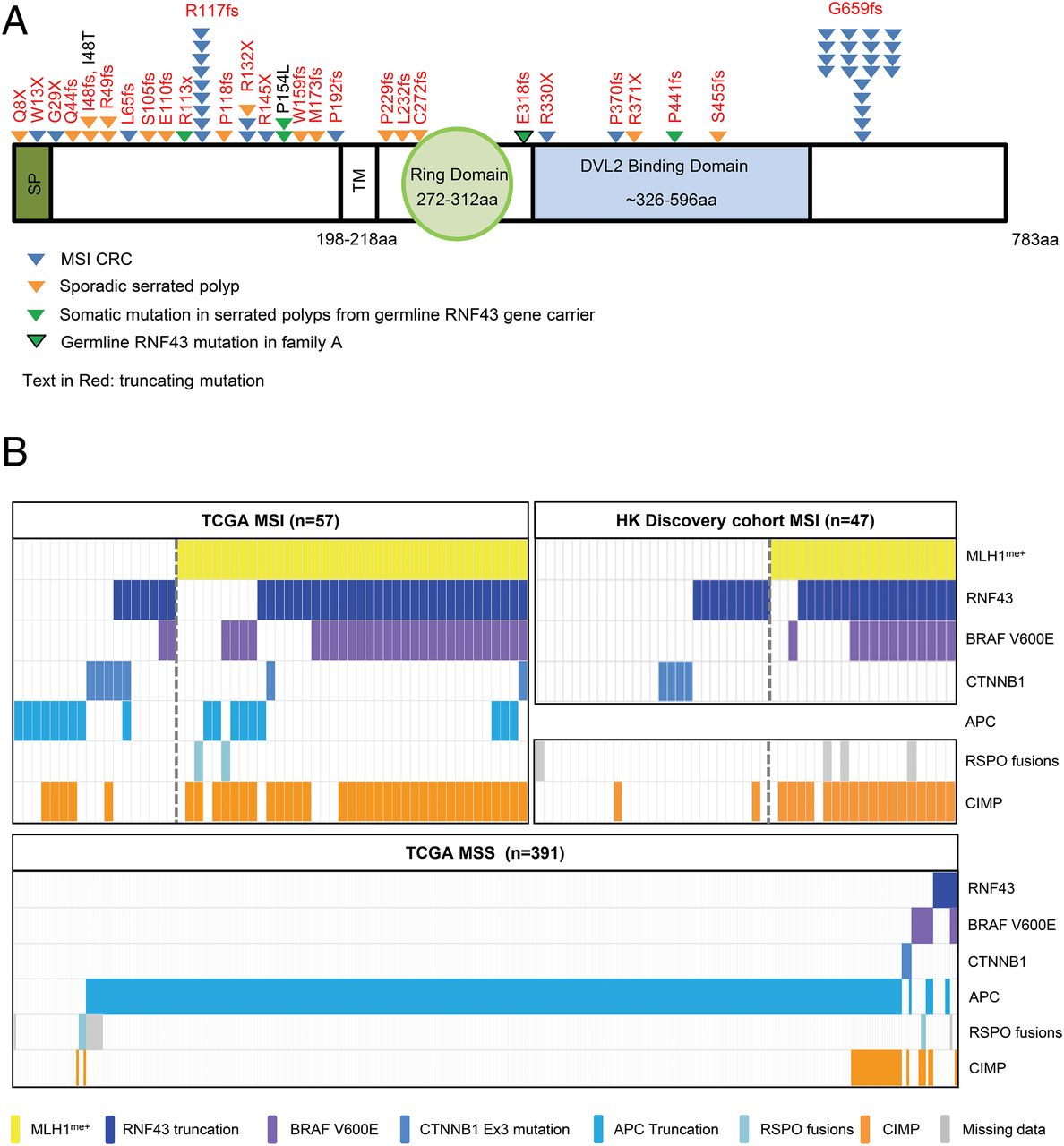
(A) Schematic diagram of germline and somatic mutations of RNF43 detected in polyps from a serrated polyposis family, a series of sporadic serrated adenomas and a series of microsatellite instability (MSI) colorectal cancers (CRCs) by Sanger sequencing. Only truncating mutations are shown for MSI CRCs. SP, signal peptides; TM, transmembrane domain. (B) Oncoplot of key oncogenic drivers in CRCs segregated into MSI with or without MLH1 promoter methylation in the Hong Kong and TCGA cohorts, and in TCGA microsatellite stable (MSS) CRCs.
Truncating mutations as potential causative genes in other patients with serrated polyposis
We did not find any gene with recurrent loss of function (LoF) mutation across the remaining three families. While we did not observe truncating mutation in other genes (ATM, PIF1, TELO2, XAF1 or RBL1) previously reported in patients with serrated polyposis,11 each of them carried several truncating mutations in other genes with functions highly relevant to cancer (see online supplementary table S3). For example, ENDOG, mutated in family B (p.R111fs), is a DNase involved in mediating caspase-independent apoptosis.21 DDX31, a RNA-binding protein mutated in family C (p.L367fs), is frequently mutated in medulloblastoma,22 and a germline truncating mutation was observed in a patient with sporadic CRC.23 Family D carries a RGS19 nonsense mutation (p.Q89X), a gene involved in regulating G-protein signalling and autophagy in intestinal cells.24 Overall, the potential significance of these genes in the causation of serrated polyposis awaits further study.
Somatic mutation of RNF43 and complete loss of wild type allele is frequent in serrated adenomas, but not in HPs
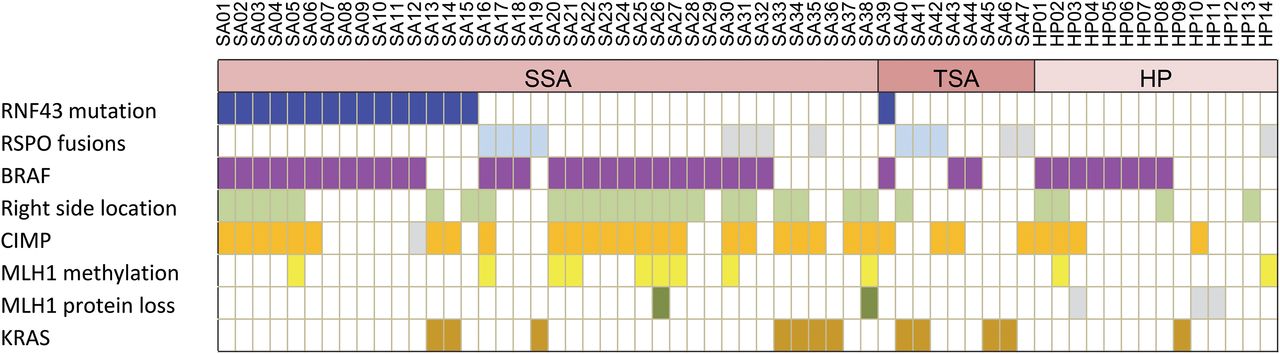
To address whether RNF43 mutation also contributes to the development of sporadic serrated polyps, we sequenced 14 HPs and 47 SSAs or TSAs. While no mutation was detected in HPs, we found RNF43 mutation in 16 SSA/TSAs (34%) and 13 had coincident BRAF mutation (p=0.112) (table 1, see online supplementary table S5, figures 3A, 4 and 5). The mutational incidence of RNF43 was significantly different between serrated adenomas and HPs (p=0.013) or conventional adenomas (p=0.002). All but one were truncating mutations, most (at least 81%) led to complete loss of wild type alleles at either the DNA or RNA level. Mutations of RNF43 in sporadic serrated adenomas did not bear an MSI signature. Immunostaining for MLH1 also detected no protein loss in these cases with RNF43 mutation. Serrated adenomas with RNF43 mutation were mostly sessile, though some showed transitional morphology to TSA, similar to those observed in germline RNF43 mutation carriers. We found no enrichment for CIMP phenotype in serrated adenomas with RNF43 mutation. CIMP phenotype was most strongly associated with right-sided serrated polyps (p<0.001), consistent with results from a previous large-scale study25 (figure 5, see online supplementary figure S4). While all serrated polyps were microsatellite stable, 11 (18%) showed methylation in the MLH1 promoter, with two associated with focal loss of MLH1 protein (figure 4B, see online supplementary figure S5C). Microdissection of the MLH1 loss region in one SSA with CIMP was performed, revealing early emergence of RNF43 hot spot frameshift mutation, preceding the onset of detectable MSI. Study of an independent series of 29 conventional adenomas revealed RNF43 mutation in one case (3.4%, p=0.002 vs SSA/TSAs) that demonstrated a distinct feature of many Paneth cells (see online supplementary table S5C). We also observed RSPO fusion in 17% of SSA/TSAs and 3.4% of conventional adenomas (see online supplementary figure S6).
Mutational incidence of RNF43 in hyperplastic polyps, sessile/traditional serrated adenomas and conventional adenomas
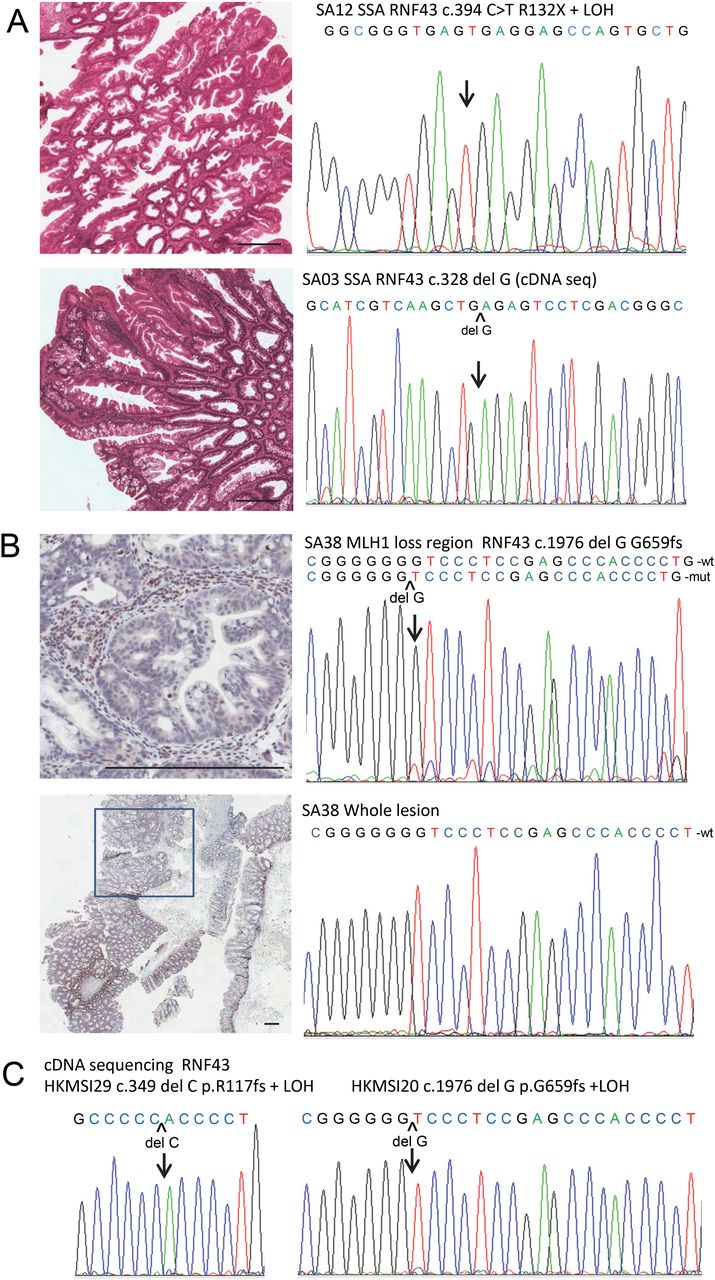
Sporadic sessile serrated adenomas (SSAs) and microsatellite instability (MSI) colorectal cancers (CRCs) with somatic RNF43 truncating mutations. (A) Two SSAs (SA12 & SA03) with truncating mutation and complete loss of wild type allele at either the DNA or RNA level. (B) A SSA (SA38) with no RNF43 mutation detected initially on whole tumour. Microdissection of the region with focal MLH1 protein loss revealed early emergence of RNF43 hot spot frameshift mutation (10% mutant fraction) in the G7 mononucleotide tract. Lower panel is low power view of lesion with box indicating the region with MLH1 protein loss. Upper panel is the corresponding high power view. Note the serrated glands with no nuclear staining, whereas the surrounding stromal cells and lymphocytes show strong staining. The right panels show the corresponding sequence chromatogram from microdissected DNA from MLH1 loss region or whole tumour. Scale bar represents 300µm. See online supplementary table S5 for a detailed mutation list and demographic data. (C) Representative sequencing chromatogram from two MSI CRCs with RNF43 mononucleotide tract hot spot mutations and complete loss of wild type allele in cDNA sequencing.

Oncoplot of RNF43 mutation and other key oncogenic changes in sporadic serrated adenomas and hyperplastic polyps (HPs). Grey box indicates missing data. CIMP, CpG island methylator phenotype; RSPO, R-spondin; SSA, sessile serrated adenoma; TSA, traditional serrated adenoma.
RNF43 mutation is significantly more frequent in MLH1 methylated MSI CRCs than MSI CRCs without MLH1 methylation
The previously reported high frequency of RNF43 mutation in MSI CRCs could be a non-specific consequence of MSI due to the presence of two short mononucleotide tracts in the gene (c.1970_1976 G7 and c.344_349 C6 tracts),16 but alternatively, it may be due to a specific and important role of RNF43 in the serrated neoplasia pathway. Thus, we examined a series of MSI CRCs (n=47) segregated into those with MLH1 methylation (n=20), which are likely evolved from serrated polyps, and those without (n=27). Complete sequencing of the RNF43 gene showed a significant difference in mutation rate between the two groups (17/20, 85% in the methylated group, vs 9/27, 33.3% in the unmethylated group, p<0.001; table 2 and figure 3). Most mutations bore the MSI signature and involved repeat tracts in the two major hot spots (c.1976delG G659fs, c.349delC R117fs), accounting for 88.5% of mutant cases, with frequent double mutations. Combined with cDNA sequencing, complete loss of wild type allele was observed in over 90% of cases with truncating mutation (figure 4C, see online supplementary table S6). Concordant with the literature, CIMP and BRAF mutation are substantially enriched in the methylated group, whereas KRAS mutation is enriched in the unmethylated group (p<0.001, table 2). To confirm the lower mutational incidence of RNF43 in MSI CRCs due to MMR gene germline mutations, we examined the two RNF43 mutational hot spots in another series of 64 paraffin MSI CRCs with confirmed germline MMR gene mutation, and noted a similar difference in mutational incidence (15/20, 75% in methylated vs 31/64, 48.4% in germline mutation group, p=0.037).
Differential mutation rates of Wnt signalling component genes and other oncogenic drivers in relation to MLH1 promoter methylation status in MSI CRCs derived from the Hong Kong (HK) and TCGA cohorts
Validation analysis of the TCGA cohort further delineated a significant differential involvement of three Wnt pathway genes between the MLH1 methylated versus unmethylated groups (RNF43 in MLH1me+; APC and CTNNB1 in MLH1me−)
The observed difference in mutational incidence suggests a different oncogenic pathway progression between the two groups of MSI CRCs. To explore this, we used TCGA exome sequencing data from 448 CRCs with linked MSI and MLH1 methylation status, and extracted the mutational incidence of key driver genes (RNF43, APC, CTNNB1, BRAF, KRAS and TP53) along with CIMP phenotype (see online supplementary table S7). We found a significant difference in the mutational incidence of all three key Wnt pathway genes between the two MSI groups (p=0.005, 0.042 and 0.027 for RNF43, APC and CTNNB1, respectively) (table 2, figure 3B). In the methylated group (n=39), RNF43 was the key gene mutated (76.9%), followed by APC (23.1%), but much less so in CTNNB1 (5.1%). In the unmethylated group (n=18), APC was the key gene mutated (50%), followed by RNF43 (38.9%) and CTNNB1 (27.8%). The differential involvement of CTNNB1 hot spot mutation, being predominantly seen in the unmethylated group, was further validated by Sanger sequencing in the Hong Kong (HK) cohorts (table 2). As expected, both BRAF mutation and CIMP were markedly enriched in the methylated group (p<0.001), whereas KRAS mutation was enriched in the unmethylated group (p=0.026). There was no difference in mutational incidence for TP53.
Co-occurrence of RNF43 and BRAF mutations in the MSI, MSS and MLH1me+ MSI CRCs
We previously noted a trend for co-occurrence of RNF43 and BRAF mutations in serrated polyps from germline RNF43 mutation carriers and in sporadic SSAs/TSAs (table 3). Similarly, we observed a significant association of RNF43 and BRAF mutations in MSI CRCs across both the HK and TCGA cohorts (p=0.003, p<0.001, respectively, table 3). To increase statistical power, we analysed the two cohorts combined. The association remained statistically significant within the MLH1 methylated CRCs, where both mutations were exceedingly common (74.5% with BRAF mutation in the RNF43 mutated group, vs 41.7% in the RNF43 wild type group, p=0.042).
Association between RNF43 truncating mutation and BRAF V600E mutation in MSI and MSS CRCs, and sporadic serrated adenomas in various study cohorts
Next, we questioned whether mutations of the two genes coexist in the MSS CRCs by interrogating the TCGA data. Although both RNF43 and BRAF mutations were uncommon (2.6% and 3.1%, respectively, see online supplementary table S7), there was statistically significant association between the two mutations (p=0.002, table 3). A similar association was also observed in a second independent data set (p=0.025).16
Similar to sporadic serrated polyps, we observed no consistent association between RNF43 mutation and the CIMP phenotype, apart from their co-occurrence in MLH1me+ CRCs. Specifically, MSS or MLH1me− CRCs with RNF43 mutation very rarely displayed CIMP (figure 3B). Mutation of RNF43 in MSS CRCs did not bear an MSI signature. Taken together, the results suggest that MSS CRCs with RNF43 mutation may have evolved from serrated adenomas with early acquisition of non-repeat tract mutation.
Organoid culture from a RNF43−/− serrated adenoma showed alleviated dependency on R-spondin
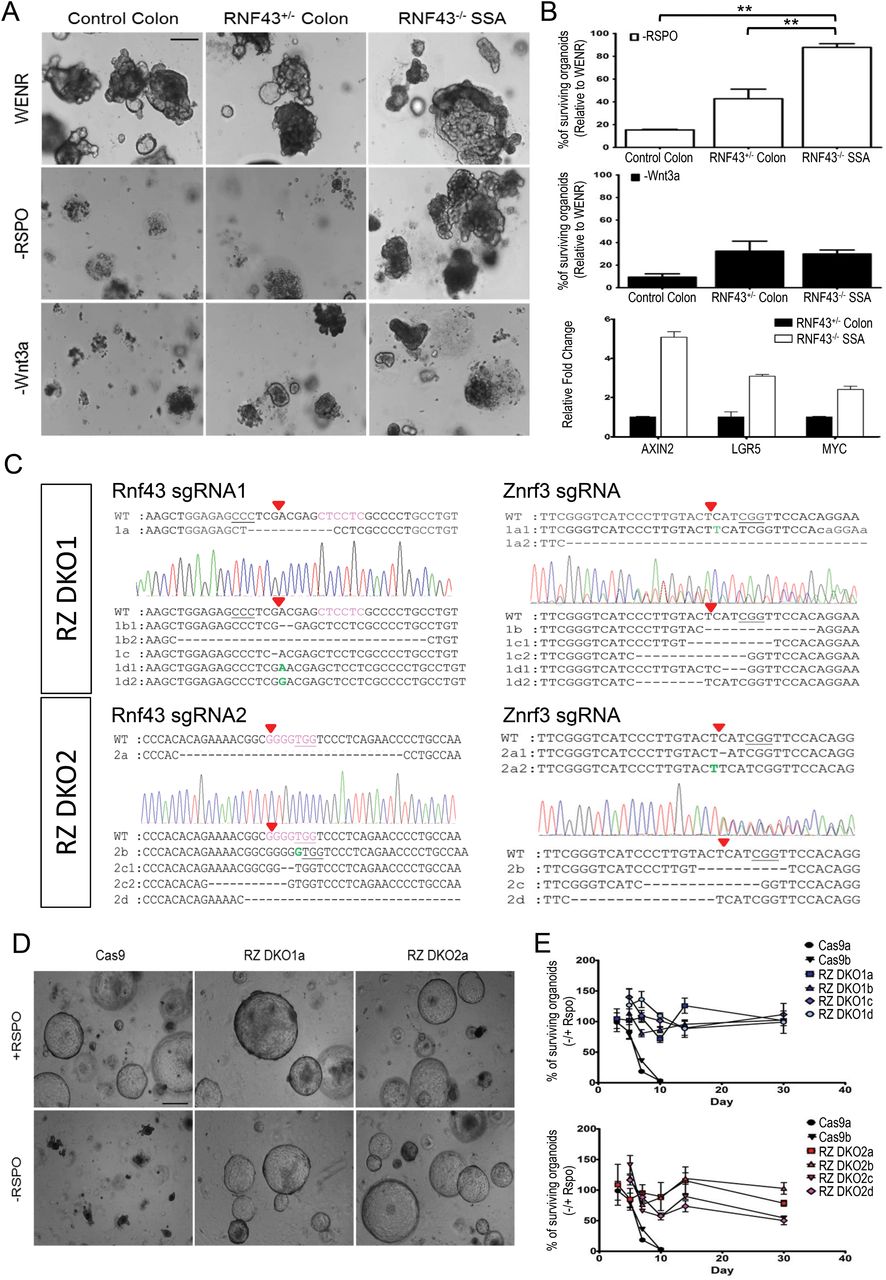
Adenomas from APC mutant mice can be grown as organoids in the absence of Wnt and R-spondin1,26 whereas those from Rnf43/Znrf3 compound mutant mice do not need Rspo1 but are dependent on Wnt.15 To understand the growth behaviour of SSAs with RNF43 mutation, we compared the RNF43−/− organoids derived from a SSA of patient A:II-3 (SP3) with her normal rectal organoids, as well as a control rectal organoid culture from another patient with CRC. Upon R-spondin1 withdrawal, the organoid death rate of the RNF43−/− SSA was significantly reduced compared with the two normal rectal organoids (figure 6A,B). While the RNF43−/− SSA organoid eventually could not survive in the long term without R-spondin1 or Wnt3a, the results suggest that RNF43 mutation may alleviate the need for R-spondin. Moreover, we confirmed the elevated expression of Wnt target genes (AXIN2, LGR5 and MYC) in the RNF43−/− SSA organoid relative to her normal rectal organoid, supporting that the second somatic hit contributes to Wnt activation.

Survival of organoids with RNF43 mutation in response to R-spondin withdrawal. (A) Bright field images of organoids derived from a sessile serrated adenoma (SSA) with RNF43 two-hit inactivation and normal colonic organoids in response to R-spondin1 withdrawal. Organoids were dissociated into single cells, seeded at 3000 cells per well in triplicate, and cultured in medium containing Wnt3a, EGF, Noggin and R-spondin1 (WENR) for comparison with medium omitting R-spondin1 or Wnt3a. Control colon, organoids derived from normal rectal mucosa from a patient with colorectal cancer (CRC); RNF43+/− colon, organoids derived from normal rectal mucosa from patient A:II-3 with germline heterozygous RNF43 mutation; RNF43−/− SSA, organoids derived from a SSA (SP3) of patient A:II-3 with complete inactivation of RNF43 through loss of heterozygosity (LOH). Scale bar represents 200 µm. (B) Upper two panels, survival of organoids scored by luminescence at day 9 (−Rspo) and day 15 (−Wnt3a), relative to WENR, with error bars representing SD. **p<0.001 by two-tailed Student's t test. The experiment was repeated twice with consistent results. Lower panel, upregulation of Wnt target genes in RNF43−/− SSA organoid by real-time qRT-PCR. (C–E) Functional characterisation of two hot spot mutations of RNF43 observed in human microsatellite instability (MSI) CRCs residing in short mononucleotide tracts (c.349delC p.R117fs and c.1976delG p.G659fs) by creating truncating mutations close to these regions in normal mouse colon organoids, along with Znrf3 truncating mutations using the Crispr/Cas9 genome editing technique. (C) Mouse genomic sequence concordant with the two human mutation hot spot regions in RNF43 are highlighted in pink. Red arrowhead denotes cleavage site; Protospacer adjacent motif (PAM) sequences are underlined. DNA sequences of eight representative clonal organoids with presence of truncating Rnf43 mutations close to the two hot spot regions along with Znrf3 truncating mutations are shown along with one representative sequence chromatogram (RZ DKO1a-d, derived from hot spot region c.349; RZ DKO2a-d, derived from hot spot region c.1976). The experiment was performed twice with clones recovered from two independent mice. Each clone had either homozygous mutations or two different truncating mutations for each of Rnf43 and Znrf3. Nucleotide in green denotes insertion and—denotes deletion. (D) Organoids carrying Rnf43−/−/Znrf3−/− double-knockout in either hot spot region survived in matrigel in the absence of R-spondin1 for at least 30 days, whereas all control organoids transduced with Cas9 only died by day 7. Scale bar represents 500 µm. (E) Graph showing the % of surviving organoids in −RSPO relative to +RSPO growth conditions, as measured by luminescence by cell-titre Glo proliferation assay at various time points. Measurements were performed in triplicate with error bars representing the SD. (See online supplementary table S9 for sgRNA sequences and supplementary table S10 for results on potential off-target sites analysis.)
RNF43 hot spot frameshift mutations induced by MSI confers R-spondin independency
Intestinal organoids from Rnf43/Znrf3 compound mutant mice were shown to survive long-term culture in the absence of Rspo1, whereas wild type organoids all died within 5 days.15 Additionally, inactivation of Rnf43 by CRISPR/Cas9, through sgRNA inducing frameshift mutations around the proximal region of the gene (amino acid 217–246), along with Znrf3 inactivation can lead to growth independent of R-spondin in mouse small-intestinal organoids.27 Thus, the functional significance of most of the proximal truncating mutations observed in the current study is unequivocal, but the most frequent hot spot mutation as a result of MSI (p.G659fs) removes the distal part of the gene where no functional domain is known. To examine if this distal hot spot frameshift mutation could attenuate RNF43 signalling, we designed two different CRISPR/Cas9 vectors to specifically target the two mononucleotide hot spots (p.R117fs and p.G659fs), with the proximal hot spot acting as a positive control, each in combination with targeting Znrf3 in mouse colon organoid culture, as single targeting of one gene failed to generate any surviving clones upon selection by puromycin and short-term R-spondin1 withdrawal. We successfully recovered clones that have two-hit inactivation of Rnf43 located adjacent to either of the hot spots, in combination with two-hit Znrf3 inactivation (figure 6C–E). These mutant clones showed continuous survival upon R-spondin withdrawal for at least a month, whereas the parallel control died by day 7. Thus, although the most common hot spot truncating mutation of RNF43 c.1976delG, G659fs, lies in the more distal part of the gene in the cytoplasmic domain, our study has confirmed that such a truncating mutation can attenuate RNF43 signalling and confer R-spondin independency. Recently, part of the intracytoplasmic domain of RNF43 has been shown to bind Dishevelled and was necessary to mediate Wnt receptor degradation.28 While the Dishevelled binding region is still proximal to the hot spot truncation, there may be as yet undiscovered functions for the C-terminal part of the gene. The functional relevance of the distal hot spot mutation is further supported by its strong negative association with APC mutation, as demonstrated previously7 and further validated in the current study (see online supplementary table S8).
Discussion
In conclusion, we have shown for the first time that RNF43 mutation constitutes an important mutated driver gene for the serrated neoplasia pathway in both the sporadic and familial polyposis settings, and its association with BRAF mutation in both MSI and MSS CRCs. Germline mutation of RNF43 accounts for 12.5% (2/16) of patients satisfying the WHO criteria of serrated polyposis syndrome in a previous study,11 and 25% in the current study. The diversity of phenotypes, in terms of number of serrated polyps observed, co-occurrence of tubular/villous adenomas and early onset CRC, as well as the early onset of serrated polyps and late age of onset of the full-blown polyposis phenotype in germline RNF43 gene carriers, as illustrated by family A, could contribute to an under-recognition of this condition and the previously unclear inheritance pattern in these families. Furthermore, another family with RNF43 truncation displaying extremely early onset serrated polyps and cancer (third decades), adds to the diversity and possibility of other environmental or genetic modifiers.12 Based on data from the Exome Aggregation Consortium (http://exac.broadinstitute.org), germline heterozygous truncating mutation of RNF43 has been observed in 0.01% of the population (covering 53 094 non-cancer individuals). Currently, patients and their first-degree relatives with serrated polyposis syndrome have a recognised substantial risk of developing CRC, but the magnitude of that risk has not been defined and colonoscopy every 1–2 years is the expert recommendation.1 ,2 A recent multicentre cohort study encompassing 434 patients with serrated polyposis syndrome recorded an incidence of CRC in 29%, with 38% having at least one first-degree relative with CRC.29 Since serrated polyposis syndrome can be caused by genes other than RNF43, based on our findings, routine germline testing for RNF43 mutation in these families should be performed. First-degree relatives from RNF43-linked families who test negative can be spared from frequent screening and a heavy psychological burden, whereas follow-up large-scale studies are needed for RNF43 gene carriers to clearly delineate their risk of cancer, so as to derive an appropriate prophylactic screening strategy.
We have also shown frequent somatic mutation of RNF43 in sporadic SSAs but not in HPs and rarely in conventional adenomas. A concurrent study also reported frequent RNF43 mutation in serrated adenomas, along with PTPRK-RSPO3 fusion, although more in TSAs than SSAs.30 Since some of the SSAs we observed in both the familial and sporadic cases showed transitional morphology to TSA with focal development of villous configuration, they may represent a morphological continuum. The malignant potential of serrated polyps has been another area of intense interest with implications on surveillance strategy.31–33 We noted an SSA with intramucosal adenocarcinoma among the SSAs with RNF43 mutation. Thus, RNF43 could constitute another useful biomarker to distinguish those with high risk of malignant progression.
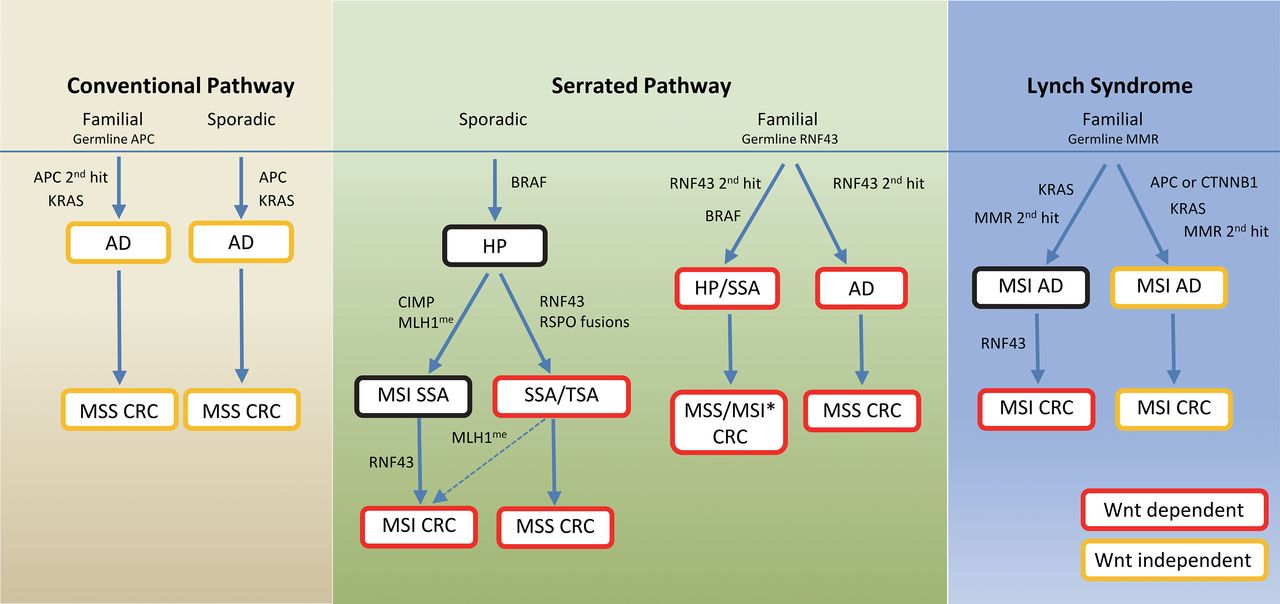
Because of the importance of Wnt signalling in colon cell homoeostasis, our study presents novel findings that show the highly prevalent, yet differential mechanisms of aberrant Wnt activation in different pathways of colon cancer development, and their relationship with other cooperating oncogenic events (figure 7). The difference in molecular pathways of evolution between the MSI and MSS CRCs has been a focus of intense study for decades, but differences between the MLH1 methylated and unmethylated groups within MSI are less clear. We now show prevalence of APC and CTNNB1 mutations in the unmethylated group and RNF43 in the methylated group. APC mutation has been shown to precede the onset of MSI in a few adenomas from germline MMR gene mutation carriers.34 Thus, the lower incidence of RNF43 mutation, despite subsequent acquisition of MSI in these adenomas, could be due to a lack of selective advantage. By contrast, we found that co-occurrence of RNF43 and BRAF mutations uniquely characterises the serrated neoplasia pathway in both the sporadic and familial forms, though there is no consistent sequential order of mutation. In sporadic serrated polyps, we found that BRAF mutation is an early event occurring in HP. Acquisition of RNF43 mutation then happens in SSA either before or after onset of MSI. We have demonstrated for the first time the early emergence of RNF43 G659fs hot spot mutation in an SSA with CIMP from a focal region with MLH1 loss, and further documented the prevalence of complete inactivation of RNF43 in most MSI CRCs involving this hot spot. Thus, CIMP tended to occur in right-sided SSAs, driving MLH1 promoter methylation/MSI/RNF43 hot spot mutation, while serrated adenomas that acquired RNF43 mutation independent of CIMP may progress predominantly to MSS CRCs or rarely MSI CRCs, as evidenced by the existence of rare cases of MLH1me+ MSI CRCs without the characteristic MSI mutational signature (eg, HKMSI2, HKMSI12). In familial cases, we observed a preferential development of serrated adenomas with BRAF mutation in germline RNF43 gene carriers, where RNF43 second-hit inactivation may precede BRAF mutation, as observed in some serrated polyps of younger gene carriers from family A (III-1 and III-2). While over 60% of the serrated polyps from family A had CIMP, we did not find MSI or MLH1 promoter methylation, and the CRC was MSS. Thus, apart from a predisposition for MSS CRC, whether germline RNF43 mutation carriers have a predisposition for MSI CRC awaits further confirmation. Oncogenic mutation may confer specific stresses that invite particular collaborating mutations, as shown by a previous study whereby induction of BRAF mutation in mouse intestines led to stem cell depletion that could be rescued by Wnt activation, providing a biological explanation for the coordinated alterations in both pathways.35 The preferential co-occurrence of RNF43, instead of APC or β-catenin, with BRAF mutation may be a consequence of complex interactions between environmental mutation load, underlying epigenetic/genetic instability driving susceptibility of specific gene loci, as well as other yet unknown factors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic summary of different pathways of colorectal carcinogenesis with differential mechanisms of Wnt and Ras-Ref-MEK-ERK signalling pathway activation. Colour of text box indicates Wnt activation status; red denotes active and Wnt dependent; orange denotes active and Wnt independent; black denotes Wnt not active. Those groups in red boxes are candidates for Wnt inhibitor treatment. AD, adenoma; HP, hyperplastic polyp; SSA, sessile serrated adenoma; TSA, traditional serrated adenoma; MSI, microsatellite instability; MSS, microsatellite stable; CRC, colorectal cancer; MMR, DNA mismatch repair genes; CIMP, CpG island methylator phenotype; MLH1me, MLH1 promoter methylation. Dashed arrow indicates a potential minor pathway. *Whether germline RNF43 mutation predisposes to MSI CRC awaits further confirmation.
Given the mutually exclusive relationship between RNF43 and APC mutation, this raises therapeutic opportunities for treatment and prevention of cancer progression in patients with sporadic, as well as familial serrated polyposis, and their derived CRCs. Specifically, many sporadic SSAs tend to be proximally located, sessile and of large size that is not amenable for endoscopic resection. Moreover, patients with sessile serrated polyposis syndrome may have an overabundance of polyps that may require total colectomy. The occurrence of germline and/or somatic RNF43 mutation in these polyps may raise a potential alternative therapeutic option through pharmacological inhibition of Wnt. Specifically, porcupine inhibitor can suppress growth of intestinal tumours from Rnf43-Znrf3 mutant mice and colon cancer organoids;36 ,37 an oral Wnt inhibitor LGK974 is currently in phase I clinical trial for cancers carrying RNF43 mutation, and has shown an in vivo effect in pancreatic cancer xenograft models with RNF43 mutation.38
Acknowledgments
The authors thank Dorothy Cheng for patient coordination, clinicians at the Hong Kong Hospital Authority for clinical care, Jonathan LK Man for technical assistance and April S Chan for editorial assistance. The authors also thank Macrogen Inc and Centre for Genomics Sciences at the University of Hong Kong for performing the exome sequencing, and the latter for other supports on Sanger sequencing and pyrosequencing. The results published here on the TCGA cohort are based upon data generated by The Cancer Genome Atlas managed by the National Cancer Institute (NCI) and National Human Genome Research Institute (NHGRI). Information about TCGA can be found at http://cancergenome.nih.gov.
References
Footnotes
HHNY and JCWL contributed equally.
Accession code Ensembl: human RNF43 mRNA, ENST00000577716.
Contributors SYL and STY conceived the study; SYL, HHNY, JCWL and SLH contributed to the project design; HHNY, JCWL, AKWC, ASYC, WYT, BCHL, SSKY and AHYM performed the experiments; JCWL performed bioinformatics analysis; WKL, WLL and JFYL were responsible for clinical care, and contributed samples, clinical data and comments on the manuscript; SLH and SYL interpreted the histology; SYL, SLH and STY were responsible for patient counselling and clinical data acquisition; HC provided support on establishment of organoid culture assay and contributed comments on the manuscript; SYL, HHNY, JCWL and SLH analysed and interpreted data; SYL, HHNY and JCWL wrote the manuscript with assistance and final approval from all coauthors.
Funding This work was supported by a donation from Mr S T Pan, the Hong Kong Cancer Fund, and the YW Kan Endowed Professorship established through a donation from the Croucher Foundation. The funders did not participate in the study design, collection, analysis and interpretation of data.
Competing interests SYL and STY have received research sponsorship from Pfizer, Merck and Servier. WKL has received honorarium for attending advisory boards of Abbie, Ferring, Ipsen and Takeda. He has also received speaker's fee from Menarini and Ferring. HC is an inventor on several patents relating to Wnt activity in cancers and a pending patent on growing organoids from patients with colorectal cancer.
Patient consent Obtained. IRB approved waivers for archival specimens.
Ethics approval Institutional review board of The University of Hong Kong/Hospital Authority Hong Kong West Cluster.
Provenance and peer review Not commissioned; externally peer reviewed.