Article Text

Abstract
Background: Patients with a multiple endocrine neoplasia type 1 (MEN1)-associated Zollinger–Ellison syndrome (ZES) show multifocal duodenal gastrinomas and precursor lesions.
Aims: To test these lesions for loss of heterozygosity (LOH) of the MEN1 gene locus on chromosome 11q13, and to investigate whether the MEN1-related endocrine cell changes also involved somatostatin cells.
Material and methods: Tissue specimens from six patients with MEN1 and ZES were analysed by immunohistochemistry and immunofluorescence. LOH analysis was performed by fluorescence in situ hybridisation (FISH), using probes containing the MEN1 gene locus and the centromere 11 (C11) region. For simultaneous analysis of hormones and allelic deletions, a combined FISH/immunofluorescence protocol was established.
Results: 28 of a total of 33 duodenal neuroendocrine tumours (NETs) were gastrin-producing tumours; 13/28 (46.4%) revealed LOH on 11q13 and/or C11. Five of the NETs were somatostatin-expressing tumours, two revealing LOH. Allelic loss was detected in tumours as small as 300 μm (gastrin) and 400 μm (somatostatin) in diameter. The gastrin-producing tumours showed different deletion/retention patterns. Hyperplastic somatostatin cell lesions, similar to those of the gastrin cells, were present in all patients. The hyperplastic lesions of both cell lines consistently retained both 11q13 alleles.
Conclusions: Allelic deletion of the MEN1 gene may reflect a pivotal event in the development of multifocal gastrin and somatostatin cell neoplasms in the duodenum of patients with MEN1. The observation of distinct deletion patterns in small synchronous tumours supports the concept that each gastrin-producing tumour in an individual MEN1 patient arises from an independent cell clone.
- C11, centromere 11
- CgA, chromogranin A
- FISH, fluorescence in situ hybridisation
- FITC, fluorescein isothiocyanate
- LOH, loss of heterozygosity
- MEN1, multiple endocrine neoplasia type 1
- NET, neuroendocrine tumour
- SSC, standard sodium citrate
- ZES, Zollinger–Ellison syndrome
Statistics from Altmetric.com
- C11, centromere 11
- CgA, chromogranin A
- FISH, fluorescence in situ hybridisation
- FITC, fluorescein isothiocyanate
- LOH, loss of heterozygosity
- MEN1, multiple endocrine neoplasia type 1
- NET, neuroendocrine tumour
- SSC, standard sodium citrate
- ZES, Zollinger–Ellison syndrome
Duodenal gastrinomas may occur sporadically or in the setting of the multiple endocrine neoplasia type 1 (MEN1) syndrome. Sporadic and MEN1-associated duodenal gastrinomas differ in their biological behaviour. Although sporadic gastrinomas are solitary tumours, MEN1-associated gastrinomas are usually multicentric.1–4
Recently, it was shown that duodenal gastrinomas in patients with MEN1 are associated with diffuse hyperplastic gastrin cell changes and multicentric gastrin-producing microtumours <1 mm in size within the grossly unaffected duodenal mucosa. It was concluded that these microscopic lesions precede the development of MEN1-associated gastrinomas.4
The vast majority of patients with MEN1 display heterozygotic germline mutations of the MEN1 gene.5,6 According to Knudson’s two-hit hypothesis for tumour suppressor genes, the development of MEN1-related tumours is associated with somatic inactivation of the wild-type MEN1 allele on chromosome 11q13.5,7,8,9,10,11,12,13,14 Recently, a loss of heterozygosity (LOH) at 11q13 was demonstrated in pancreatic microadenomas of patients with MEN1 and interpreted as a very early event in MEN1-related pancreatic tumorigenesis.15 We do not know at present at what stage a similar change occurs in MEN1-related duodenal neuroendocrine tumours (NETs). If deletion of the wild-type allele of the MEN1 gene is also an early step in gastrinoma development, LOH would represent a decisive molecular event discriminating between hyperplastic and early neoplastic lesions.
To date, PCR-based LOH analysis and fluorescence in situ hybridisation (FISH) analysis of macrotumours have been used to examine defined chromosome loci in hereditary and non-hereditary duodenopancreatic NETs.9,12,14,16–19 However, these techniques are less than optimal for analysing scattered hormone-expressing cells—for example, in the diffuse endocrine system of the gut.
To analyse the occurrence and frequency of LOH on chromosome 11q13 in duodenal MEN1-related microtumours, hyperplastic lesions and duodenal neuroendocrine cells, we established a combined FISH/immunofluorescence method, enabling us to analyse allelic deletions and hormone expression in duodenal neuroendocrine cells at the same time. In addition to LOH analysis, we examined whether the MEN1-related changes in the duodenum are restricted to the gastrin cell lineage or additionally involve somatostatin cells, because sporadic somatostatin-producing tumours are second in frequency among all NETs of the duodenum,20,21 and have been described in association with the MEN1 syndrome.22
MATERIALS AND METHODS
Patients
The study included six patients with MEN1 having duodenal gastrinomas and Zollinger–Ellison syndrome (ZES; three men and three women; mean age 43 years; range 33–54 years at the time of operation), whose specimens were collected between 1980 and 2005 in the consultation files and NET archives of the Departments of Pathology of the Universities of Kiel (Kiel, Germany) and Zürich (Zürich, Switzerland). Four of these patients were included in earlier studies investigating the histopathology of duodenal gastrinomas and pancreatic endocrine tumours in MEN1.1,4,23 The diagnosis of ZES was established on the basis of clinical symptoms: an increased fasting serum gastrin level (>125 pg/ml) combined with a low pH in the fluid of the stomach and a positive secretin-stimulation test (increase of serum gastrin concentration to >200 pg/ml). Since none of the patients had a somatostatinoma syndrome, SOM levels were not determined. All patients fulfilled the clinical criteria of an MEN1 syndrome—that is, at least two MEN1-related endocrine tumours or one of these tumours and a first-degree relative with MEN1 (table 1). An MEN1 germline mutation was proven in four patients. Tissue samples from all regions of the duodenum of three patients who had a Whipple resection for pancreatic ductal adenocarcinoma (three men; mean age 53 years; range 39–69 years) and from three patients who had sporadic (non-MEN1-associated) gastrinomas (two men and one woman; mean age 60 years; range 56–72 years) were used as control tissues.
Clinicopathological features and loss of heterozygosity analysis of neuroendocrine tumours and precursor lesions
Tumour classification
Paraffin wax-embedded tissue blocks containing tumour and extra-tumorous duodenal mucosa fixed in 4% formaldehyde were examined from each patient. A neuroendocrine neoplasm was identified by its positive staining for the markers synaptophysin and chromogranin. Endocrine tumours >2 mm were referred to as macrotumours and tumours from 250 μm to 2 mm as microtumours, as previously proposed.4 Tumours expressing gastrin and associated with a ZES were termed gastrinomas. However, in the case of multicentric tumours, it cannot be determined which one was responsible for the ZES. Therefore, we use the term “gastrin-producing tumour” instead of “gastrinoma”. Hyperplastic lesions were classified as simple, linear, micronodular or macronodular neuroendocrine cell hyperplasia.4 Two or more duodenal NETs (microadenoma and/or macrotumour) were defined as multifocal tumours.1
Immunohistochemistry
Sections (3–4 μm) were cut at 100 μm intervals from paraffin wax-embedded tissue blocks. Serial sections were immunostained with the following antibodies: chromogranin A (CgA; mouse monoclonal, dilution 1:40 and 1:4 for immunofluorescence, BioGenex, San Ramon, California, USA), synaptophysin (mouse monoclonal, dilution 1:100 and 1:10 for immunofluorescence, HyTest, Turku, Finland, and rabbit, polyclonal, dilution 1:50, Dako, Glostrup, Denmark), SOM (rabbit, polyclonal, dilution 1:200 and 1:50 for immunofluorescence, Dako, Hamburg, Germany, and rat monoclonal, dilution 1:100 for immunofluorescence, Acris, Hiddenhausen, Germany), gastrin (rabbit, polyclonal, dilution 1:3000 and 1:200 for immunofluorescence, Paesel, Frankfurt, Germany, and mouse monoclonal, dilution 1:50 for immunofluorescence, Immunotech, Marseille, France) and Ki-67 (mouse monoclonal, dilution 1:1, Department of Pathology, Kiel, Germany). Deparaffinised 3–4 μm thick sections were rehydrated and subjected to heat-induced epitope retrieval procedures.24,25 Before application of the primary antibody, blocking with non-immune serum was performed for 20 min. After incubation for 45 min, the reaction was detected with species-specific biotinylated secondary antibodies (Dianova, Hamburg, Germany) for 45 min, washed in phosphate-buffered saline and incubated for 30 min with the avidin–biotin–peroxidase complex reagents (Vectastatin Elite ABC Kit, Boehringer, Ingelheim, Germany). Immunoreactions were visualised with 3′,3-diaminobenzidine (Sigma, Deisenhofen, Germany). Immunostained sections were analysed and photographed with an Axioskop 50 microscope (Zeiss, Oberkochen, Germany).
Assessment of non-tumourous gastrin and somatostatin lesions
The non-tumorous endocrine cell lesions were classified as proposed previously.4 At least 10 serial sections at a distance of >0.5 cm from the tumour were assessed. Morphometric analysis to analyse the endocrine cell density was performed with an occulometer (Netzmicron, Carl Zeiss, Jena, Germany), as described previously.4,26,27 Data are given as mean (SD).
Double fluorescence microscopy
Double fluorescence microscopy on serial sections was used to analyse the expression of general neuroendocrine markers (synaptophysin and CgA) in relation to cells staining for gastrin and somatostatin. For this, the sections were covered with a mixture of the relevant primary antibodies in appropriate dilutions. Before antibody application, the tissue sections were incubated in 4× standard sodium citrate (SSC)/5% non-fat dry milk for 10 min and 4× SSC/0.05% Tween to reduce autofluorescence and to block unspecific binding sites. The slides were incubated with the primary antibodies for 2 h and then washed in 4× SSC/0.05% Tween. Subsequently, the slides were incubated with the secondary antibodies for 1 h at 37°C (Alexa 488 anti-mouse, 1:1000, Alexa 488 anti-rabbit, 1:1000, Alexa 488 anti-rat 1:1000, all Molecular Probes, Eugene, Oregon, USA, and Cy3 anti-mouse 1:1000, Cy3 anti-rabbit, 1:1000, Cy 3 anti-rat 1:1000, all Dianova, Hamburg, Germany). The sections were counterstained for nuclei using Hoechst A33528 dye at a concentration of 0.025 mg/ml and extensively washed in 4× SSC/0.05% Tween. Then the sections were mounted using ProLong anti-fade mounting medium (Molecular Probes). For the double fluorescence analysis an Olympus BX 61 fluorescence microscope (Olympus, Hamburg, Germany) was used.
LOH analysis of chromosomal region 11q13 and centromere 11
Double-target FISH was performed in all tumours using a chromosome 11-specific centromere probe (pLC11A) in combination with an 11q13-specific probe (RP1CTD-2220I9) containing the MEN1 gene. Both FISH probes were labelled with fluorescein-12-dUTP (spectrum green) and biotin by nick translation (Boehringer, Mannheim, Germany) and precipitated in ethanol in the presence of 50×Cot1-DNA, sodium acetate and glycogen. The DNA pellet was resuspended in the hybridisation buffer (50% formamide, 10% dextran sulphate, 2× SSC) with a final concentration of 250 ng probe/10 μl. After dehydration in an ethanol series and blocking for 30 min in 1% methanolic H2O2, the slides were denaturated in 0.2 M HCl at room temperature for 20 min and in 1 M NaSCN for 30 min at 80°C. Proteinase treatment was performed by incubation in 0.4 g/l pepsin in 10 mM TRIS HCl solution for 10 min at 37°C, followed by post-fixation in 4% formalin for 10 min at room temperature. After sequential washings in 2× SSC and distilled water, both probes were applied to the tissue specimens, denaturated at 80°C for 5 min and then incubated at 37°C overnight in a humidified chamber. Post-hybridisation washes were performed at 45°C in 50% formamide/2× SSC (two times for 5 min). The hybridised fluorescein-labelled probes were detected using a cascade of rabbit anti-fluorescein isothiocyanate (anti-FITC) immunoglobulins (1:1000, Dako), FITC-conjugated swine anti-rabbit immunoglobulins (1:100, Dako) and FITC-conjugated rabbit anti-swine immunoglobulins (1:1000, Dako). The biotin-labelled probes were detected using a cascade of avidin–tetramethylrhodamine isothiocyanate (1:100, Vector, Burlingame, California, USA), biotinylated anti-avidin D (1:100) and avidin–tetramethylrhodamine isothiocyanate (1:100). The slides were washed in 4× SSC/0.05% Tween, air dried and then mounted in Vectashild (Vector) containing 0.5 μg/ml of 4′,6-diamidino-2-phenylindole-antifade (Sigma) for nuclear counterstaining. Images were recorded with Analysis software (Olympus Biosystems, Hamburg, Germany).
Simultaneous FISH and hormone expression analysis
The specificity and sensitivity of simultaneous FISH/immunofluorescence detection of endocrine cells was first tested on normal duodenal mucosa from the three controls and then applied to the patients’ tissue specimens. The sections were deparafinised in xylene, washed in an ethanol series and rehydrated in 4× SSC/Tween for 10 min and then pretreated overnight in a 4× SSC/5% non-fat dry milk solution at 37°C. The denaturation, hybridisation and post-hybridisation were performed as described above. The 11q13 signal was amplified by a cascade of rabbit anti-FITC immunoglobulins (1:1000), FITC-conjugated swine anti-rabbit immunoglobulins (1:100) and FITC-conjugated rabbit anti-swine immunoglobulins (1:1000). Primary antibodies against synaptophysin, gastrin or somatostatin were applied in the second step of the detection cascade in the final dilutions described above, and incubated for 45 min. After washing in 4× SSC/0.05% Tween, the slides were incubated with the secondary antibodies for 1 h at 37°C (Alexa 488 anti-mouse, 1:1000, Alexa 488 anti-rabbit, 1:1000, Alexa 488 anti-rat 1:1000) together with the FITC-conjugated rabbit anti-swine immunoglobulins. Counterstaining and microscopic analysis were performed as described above.
Specificity controls and evaluation
The specificity controls of the FISH and the immunostainings included: (1) omission of individual steps of the detection cascade; (2) performance of the FISH/immunohistochemistry protocol without FISH probes or (3) without primary antibodies; and (4) scanning and digitalisation of immunohistochemically stained sections and comparison with subsequently obtained FISH results on the same sections.
For comparative evaluation of histological, immunohistochemical and LOH data, multiple sets of four serial sections (3 μm thick) of each tissue specimen were cut and stained in the following order: H&E, synaptophysin, gastrin or somatostatin and simultaneous FISH for 11q13 (red) and C11 (green). The presence of neuroendocrine cell lesions with only one probe signal in >80% of the cells was interpreted as an allelic deletion. Normal duodenal tissue specimens or connective tissue in the vicinity of the lesions served as internal controls. They exhibited nuclei with two probe signals at a frequency of >80%.
Statistical evaluation
The size of lesions and LOH results were analysed for independence by the χ2 test. Differences were considered significant at p<0.01.
Ethics
Anonymised archival material was used in this study, which was approved according to the guidelines of the local ethics committee (D430/2005).
RESULTS
Gastrin and somatostatin cell neoplasms
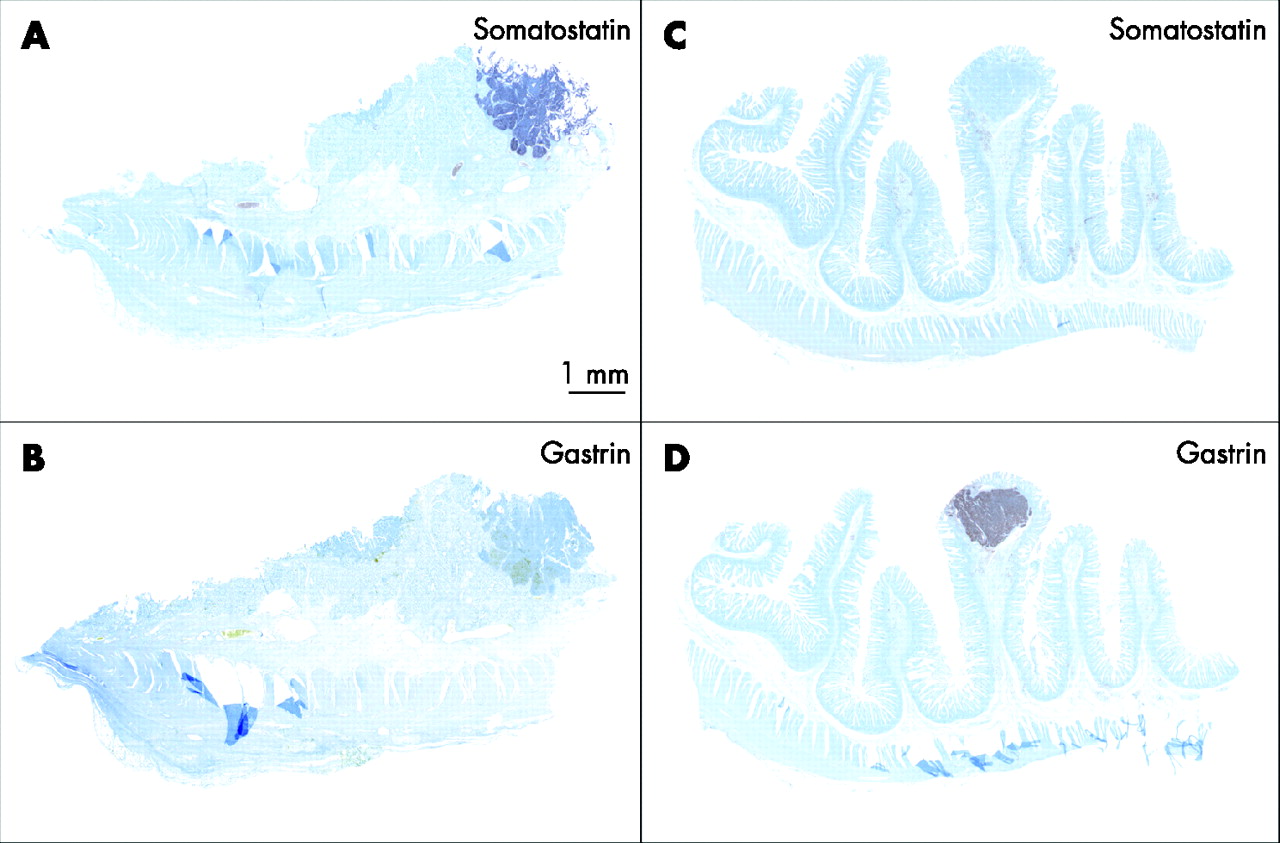
Table 1 summarises the clinicopathological and LOH data on the NETs and their associated precursor lesions in six patients. A total of 33 NETs were detected (mean diameter 4 (3) mm, range 0.3–20 mm), most of them (23/33; 70%) being microtumours with a diameter of 300 μm to 2 mm. Even when tiny, with a diameter of <1 mm, their growth pattern consisted of solid nests or trabecular cords of monomorphous cells, surrounded by thick collagen bands, fulfilling the criteria of microtumours (figs 1 and 2A). The vast majority of these tumours were localised within the Brunner’s glands (26/33; 79%). In addition to gastrin-expressing NETs, five somatostatin-expressing tumours were found in three individuals. They accounted for 15% of all MEN1-related duodenal NETs (table 1). Their diameters and histological pattern did not differ from those of the gastrin-producing tumours (fig 1 and table 1). There were no NETs without either gastrin or somatostatin expression.

Multiple endocrine neoplasia type 1-associated duodenal somatostatin- and gastrin-producing microtumours <2 mm in diameter.

Loss of heterozygosity (LOH) analysis of chromosome 11q13 (red) and centromere 11 (C11; green) in duodenal microtumours (adjacent sections). (A, B) Somatostatin-immunoreactive microtumour within the Brunner’s glands revealing LOH for both chromosomal regions. (C–D) Gastrin-producing microtumour in close association with the mucosa showing LOH for 11q13 but not for C11. (E–F) gastrin-producing microtumour within the submucosa revealing no LOH for either allele.
FISH analysis of chromosome 11q13 and the centromeric region performed on adjacent sections revealed three distinct patterns: (1) allelic deletion of both loci (3/33 tumours, 9%; fig 2A,B); (2) allelic deletion of the MEN1 locus only with retention of both C11 (12/33 tumours, 36%; fig 2C,D); and (3) retention of heterozygosity for both markers (18/33 tumours, 55%; fig 2E,F).
In five patients different deletion/retention patterns were seen in synchronous tumours that looked histologically similar (table 1). Chromosomal gains or homozygotic deletions were not detected. The diameters of the tiniest gastrin and somatostatin producing neoplastic lesions revealing LOH on chromosome 11q13 were 0.3 and 0.4 mm, respectively (table 1). A comparison of the sizes of the tumours with and without allelic deletions failed to reveal a significant difference (p = 0.25).
Hyperplastic gastrin and somatostatin cell lesions
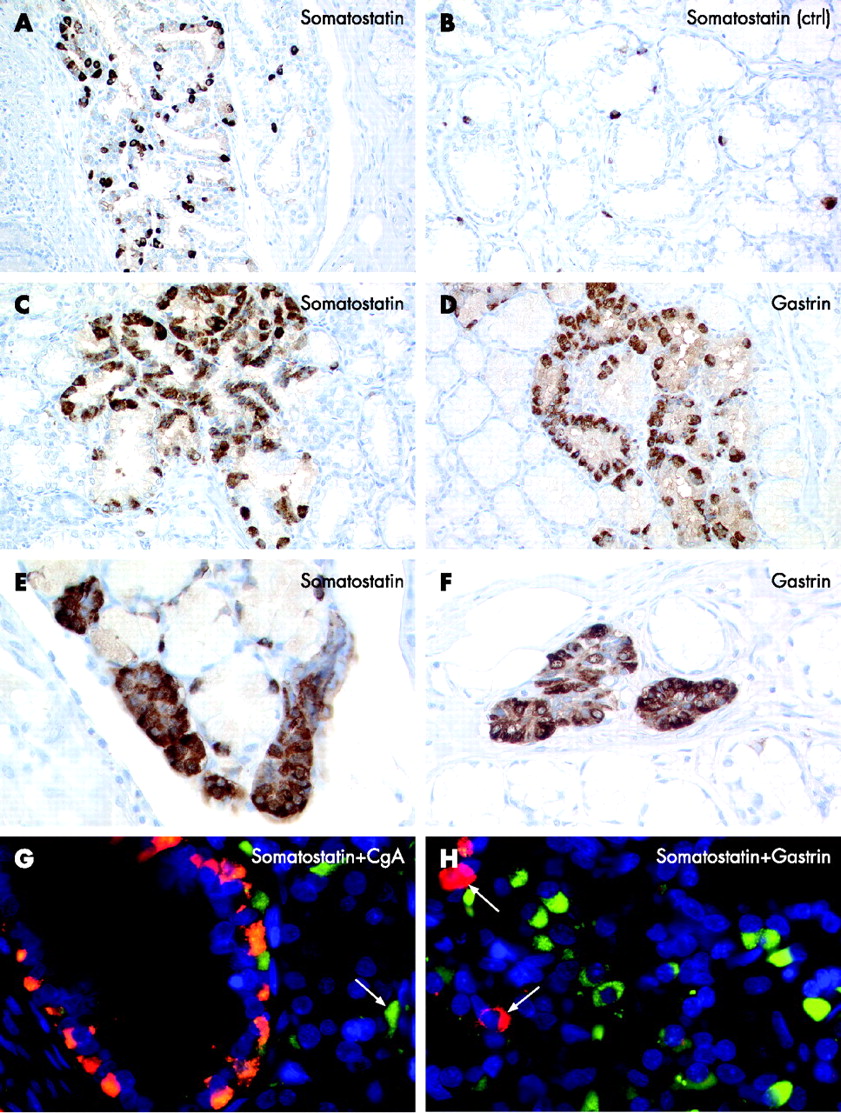
Proliferative gastrin and somatostatin cell lesions were present in all six patients (table 1). The hyperplastic changes were mainly localised within the Brunner’s glands and the mucosal–submucosal junction. Morphometric analysis and careful examination of control specimens confirmed a diffuse increase in gastrin and somatostatin cells (fig 3A,B). Somatostatin cell lesions were less frequent and less distinct than gastrin cell lesions. They consisted of diffuse (6/6 somatostatin, 6/6 gastrin; fig 3A), linear (4/6 somatostatin, 6/6 gastrin; fig 3C,D) or micronodular hyperplasia (3/6 somatostatin, 5/6 gastrin; fig 3E,F). In addition, two patients showed a total of five enlarged cell nodules with a mean (SD) diameter of 115 (17) μm. These showed gastrin, but no somatostatin expression. Double fluorescence analysis revealed mutually exclusive expression of gastrin or somatostatin in areas of endocrine cell hyperplasia. In addition, endocrine cells in these areas showed reduced contents of CgA (fig 3G,H). Double immunofluorescence using synaptophysin combined with somatostatin or gastrin on serial sections stained all hyperplastic lesions for either one hormone or the other (fig 3G). Application of fluorochrome-enhanced secondary antibodies directed against the primary peptide antibodies during the last step of the FISH detection cascade resulted in specific and sensitive simultaneous visualisation of chromosomal loci (11q13 and C11) and synaptophysin, CgA, gastrin or somatostatin. Duodenal neuroendocrine cells from control specimens, which stained for gastrin, somatostatin or CgA, consistently showed two chromosomal signals in >80% of all cells. Analysis of the duodenal specimens from the patients with MEN1 consistently revealed retention of heterozygosity on chromosome 11q13 and C11 in normal-appearing endocrine cells as well as in CgA, gastrin or somatostatin-expressing cells showing diffuse, linear or nodular hyperplasia (table 1 and fig 4).

Comparative analysis of duodenal somatostatin and gastrin cell precursor lesions. (A, B) Diffuse hyperplasia of somatostatin cells (A) within the Brunner’s glands compared with the control specimen (ctrl; B). (C–F) Chain-forming and nodular hyperplasia of somatostatin cells (C, E) resembling hyperplastic gastrin cell lesions (D, F). (G) Double fluorescence microscopy (composite image) revealing a marked loss of chromogranin A (CgA; green; arrow, single endocrine cell) in an area of diffuse and linear somatostatin cell hyperplasia (red). Faint yellow staining in a few somatostatin cells indicates low expression levels of CgA. (H) Cellular segregation of somatostatin (red, arrows) and gastrin (green) cells in an area with diffuse gastrin cell hyperplasia.
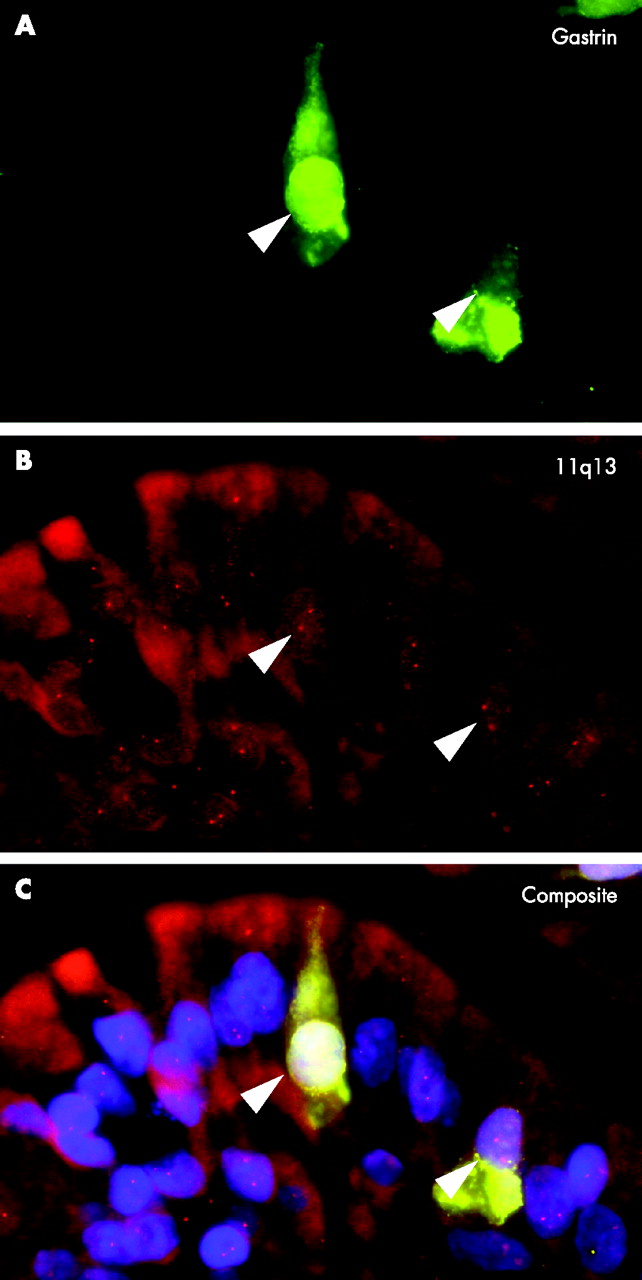
{kind=link}
{kind=link}
{kind=link}
{kind=link}
. Loss of heterozygosity (LOH) analysis of gastrin cell hyperplasia. Simultaneous detection of gastrin (green in A and C) and chromosome 11q13 (red in B and C) in an area of diffuse gastrin cell hyperplasia revealing no LOH of chromosome 11q13.
DISCUSSION
We have shown previously that MEN1-associated duodenal gastrin-producing tumours are associated with presumable precursor lesions—that is, hyperplastic gastrin cell proliferations. On the basis of morphology, we discriminated macrotumours (>2 mm) from microtumours (0.25–2 mm) and hyperplastic lesions.4 As LOH of the wild-type MEN1 allele is an early step in other MEN1-associated tumours, we examined gastrin-producing macrotumours, microtumours and hyperplastic lesions for MEN1 LOH. To detect simultaneously the allelic deletion on chromosome 11q13 at the tissue level and gastrin production, we established a combined FISH/immunofluorescence protocol permitting the assessment of LOH in neuroendocrine cells defined by their peptide hormone expression. This method permits a more reliable identification and in situ analysis of small endocrine cell lesions and single endocrine cells than PCR-based analysis of microdissected tissue samples and conventional FISH analysis. By using this technique, we were able to demonstrate allelic deletions on chromosome 11 in MEN1-associated duodenal microtumours. By contrast, hyperplastic GAS cell proliferations did not show allelic deletions, thus confirming the nomenclature based on morphology.
In 28 MEN1-associated duodenal gastrin-producing tumours in six patients, we found an LOH rate of 46%. This result is comparable to the 41% reported in MEN1-associated duodenal gastrinomas (all macrotumours) in a study using PCR-based technology.11 The smallest gastrin cell tumour in our series revealing LOH had a diameter of 300 μm. This finding provides strong evidence that these small cell nodules indeed represent early neoplasms, as they show a clonal loss of one MEN1 allele. Regardless of the tumour’s site or size, three distinct deletion patterns could be observed: (1) an allelic deletion of 11q13 and C11; (2) an allelic deletion of the MEN1 gene locus with retention of both C11 signals; or (3) no deletions at either locus in synchronous tumours. These findings support the concept that each gastrin-producing tumour in an individual patient with MEN1 arises from an independent second hit, leading to a loss of the functional MEN1 allele. Of the 13 gastrin-producing tumours with LOH on chromosome 11q13, four additionally displayed LOH of the centromeric region detected by the marker C11. This finding indicates a loss of the entire chromosome 11, which is known to occur in MEN1-associated endocrine tumours.14 Our finding that 55% of the duodenal MEN1-associated tumours lacked a deletion at the MEN1 gene locus is intriguing at first glance. However, it reflects the frequency of allelic loss reported in duodenal macrogastrinomas.11 Since inactivation of the wild-type MEN1 allele due to small intragenic somatic mutations is responsible for the development of macrotumours,28 the same mechanism may be present in the microtumours. Whether the loss of the wild-type MEN1 allele alone is sufficient to lead to tumour formation is not yet known. However, it is possible that this genetic alteration is a decisive step, or at least one of the important steps, in the development of duodenal gastrinomas in patients with MEN1.
In contrast with tumours, the foci of hyperplastic duodenal gastrin cells described as simple, linear or nodular hyperplasia consistently lacked LOH on chromosome 11q13. This finding suggests that although these gastrin cells carry the MEN1 germline mutation, they are hyperplastic but not neoplastic. The mechanisms leading to this hyperplasia are still unclear.
It is well established that multifocal duodenal gastrinomas develop in patients with MEN1.1,5,23,29,30 Three patients in our series developed, in addition to gastrinomas, a total of five somatostatin-expressing tumours (one macrotumour and four microtumours), whereas foci of hyperplastic somatostatin cells were present in all patients with MEN1. The reason why these somatostatin cell lesions and tiny neoplasms were overlooked in previous studies may be the lesser extent of the hyperplastic and neoplastic somatostatin cell changes than that of gastrin cells. In addition, as none of the patients had evidence of a somatostatinoma syndrome, these tumours were not expected.
In MEN1, it has been demonstrated that all four established pancreatic islet cell types are involved in tumour formation, although to varying extents.23,29,31 By using double fluorescence microscopy and combined FISH/immunofluorescence analysis, we showed in the duodenum that (1) gastrin and somatostatin cell lesions were clearly separated from each other; (2) all tumours expressing the general neuroendocrine markers CgA/synaptophysin were gastrin or somatostatin positive; (3) the hyperplastic lesions stained either for gastrin (frequently) or somatostatin (rarely); and (4) the hyperplastic somatostatin cells, like the hyperplastic gastrin cells, retained both 11q13 alleles, whereas the neoplastic somatostatin cells showed LOH at 11q13. We reported previously the absence of hyperplastic or neoplastic endocrine lesions expressing the duodenal hormones cholecystokinin, motilin, secretin and gastric inhibitory peptide.4 It remains to be investigated why gastrin cells and somatostatin cells are more sensitive to proliferative stimuli than other duodenal endocrine cell types in the context of the MEN1 syndrome.
In summary, we have shown that the hyperplastic gastrin and somatostatin cells that precede tumour development in the MEN1 duodenum retain both MEN1 alleles, while half of the micro- and macrotumours have lost one allele. From this finding, we conclude that allelic deletion of the MEN1 gene may reflect one decisive initial event in the development of multifocal gastrin or somatostatin cell neoplasms in the duodenum of patients with MEN1. Multifocal duodenal endocrine tumours presumably arise from independent clonal events subsequent to the germline MEN1 mutation.
Acknowledgments
We thank Maike Pacena, Anja Bredtmann, Sonja Schmid, Marion Bawohl, Franziska Nötzli, Klaus Schönheinz and Antoniella Santuccione for their excellent technical assistance. We are indebted to Wolfram Jochum and Ernst-Jan Speel for their help in establishing the protocols, Katherine Dege for critically reading the manuscript and Holger Moch for his extensive support.
REFERENCES
Footnotes
-
↵* These authors contributed equally to this work.
-
Published Online First 27 January 2007
-
Funding: This study was supported by the Hensel Stiftung Kiel (F370011, MA and GK), the Swiss National Foundation (SNF 31-18257, AP and PK) and the German Society of Pathology (MA). Some results of this study are part of NG’s MD thesis. NG and TH have research fellowships sponsored by Ipsen, Ettlingen and the Hensel Stiftung, Kiel, Germany.
-
Competing interests: None.