






Abstract
Aims: Results of several animal studies suggest that similar to humans, female rodents are more susceptible to chronic alcohol-induced liver disease (ALD). The aim of the present study was to determine whether female mice are more susceptible to acute alcohol-induced steatosis than male mice and to investigate possible mechanisms involved. Methods: Male and female C57BL/6J mice received one single dose of ethanol (6 g/kg bodyweight) or isocaloric maltose-dextrin solution intragastrically. Plasma alcohol concentration, markers of hepatic steatosis, activation of the TLR-4 signaling cascade and triglyceride export as well as lipid peroxidation and of iron metabolism were measured 12 h after acute alcohol intake. Results: In male and female ethanol-treated mice, plasma alcohol concentrations were still markedly increased 12 h after the alcohol challenge, which was associated with a significant accumulation of lipids in the liver and increase of transaminases in plasma; however, lipid accumulation was ∼3-fold higher in females in comparison with male animals. Expression of MyD88 was only found to be significantly induced in livers of female alcohol-exposed mice, whereas protein levels of ApoB were found to be significantly lower only in livers of female mice exposed to ethanol. Levels of 4-HNE protein adducts and ferritin were induced in livers of male and female ethanol-treated mice. Conclusion: Taken together, these data suggest that female mice are also more susceptible to acute alcohol-induced liver steatosis and that this involves an increased activation of TLR-4-dependent signaling pathways in the liver.
INTRODUCTION
Despite intense research efforts, mechanisms involved in the onset but also progression of alcohol-induced liver diseases (ALD) have not yet been fully understood. Results of human but also animal studies suggest that besides the hypermetabolic state resulting from a shift in the ratio of NAD+ to NADH+H+, ethanol intake, even when consumed acutely, is associated with impairments of the intestinal barrier function (e.g. damage of villi in the small intestine) (Bode et al., 1991; Bode and Bode, 1992; Parlesak et al., 2000), a more fecal type of microbiota in the small intestine (Bode, 1980) and increased permeation of intestinal bacterial endotoxin (Bode et al., 1987) subsequently leading to an activation of Kupffer cells and herein particularly Toll-like receptor (TLR)-4-dependent signaling cascades. TLR4 can activate different signaling pathways via recruitment of the interferon regulatory factor (IRF)-3 (Palsson-McDermott and O'Neill, 2004), or the common TLR adaptor, myeloid differentiation factor 88 (MyD88), which has been shown to play a crucial role in the development of acute alcohol-induced steatosis (Kanuri et al., 2009). In recent years, it has further been shown that similar to chronic alcohol ingestion acute alcohol exposure in mice leads to tumor necrosis factor receptor (TNFR) 1-dependent induction of plasminogen activator inhibitor (PAI)-1, which in turn may affect hepatic lipid export mediated through microsomal triglyceride transfer protein (MTP)-dependent signaling cascades (Sugimoto et al., 2002; Tomita et al., 2004; Bergheim et al., 2006a).
Results of several studies further suggest that alcohol and iron interact synergistically and cause liver injury. Indeed, it has been shown that iron increases the hepatotoxicity caused by alcohol in rodents (Stal and Hultcrantz, 1993) and that alcoholic liver diseases are often associated with an iron overload (Kohgo et al., 2008). Furthermore, Tsukamoto et al. (1999) showed that iron primes hepatic macrophages for NFκB activation in alcoholic liver injury. In support of these findings, She et al. (2002) found that iron serves as a direct agonist to activate IKK, NFκB and TNFα promoter activity and to induce the release of TNFα protein in cultured Kupffer cells in a redox status-dependent manner; however, if alterations in iron status or metabolism contribute also to the change found in the liver after acute alcohol ingestion (e.g. lipid accumulation) remains to be determined.
Results of several human studies suggest that women have an increased susceptibility to alcoholic liver diseases compared with men (for overview see Eagon (2010)). Indeed, it has been shown that only half of the averagely consumed amount of alcohol is sufficient to cause comparable damage to health in women (Becker et al., 1996). Furthermore, at time of diagnosis, liver damage is more advanced in female alcohol abusers, despite a reported shorter history of alcohol abuse and a lower intake of alcohol in comparison with men (Wilkinson et al., 1969; Ashley et al., 1977; Morgan and Sherlock, 1977; Levi and Chalmers, 1978; Saunders et al., 1981; Tuyns and Pequignot, 1984; Pares et al., 1986; Marbet et al., 1987; Bouchier et al., 1992). These clinical findings are also supported by results of several animal studies. For instance, it has been shown in an intragastric model of chronic alcohol feeding that female rats exhibit greater susceptibility to ALD than male animals (Iimuro et al., 1997) and that the gonadal hormone estrogen is involved in the development of early ALD (Yin et al., 2000). Indeed, results of animal studies suggest that estrogen may be critical for the higher susceptibility of female mice to alcohol (Ikejima et al., 1998; Yin et al., 2000). For instance, Yin et al. (2000) reported an increased gut permeability and higher plasma endotoxin levels in the portal vein after chronic intake of alcohol in female rats. Furthermore, Ikejima et al. (1998) found in their animal studies that CD14 expression on Kupffer cells and endotoxin sensitivity of Kupffer cells towards lipopolysaccharide (LPS) was markedly elevated in female estrogen-treated rats; however, mechanisms involved in these sex-specific differences and herein the role of estrogen are still poorly understood.
However, whether female mice are also more susceptible to acute alcohol-induced liver steatosis and if so, whether this also involves alterations at the level of intestinal permeability and Kupffer cell activation or alterations of iron metabolism has not yet been clarified. Starting from this background, the aim of the present study was to test whether female mice are more susceptible to acute alcohol-induced fatty liver compared with male mice and if so, whether changes in Kupffer cell activation, hepatic lipid export (e.g. MTP activity), and iron metabolism are involved in the greater susceptibility of female mice to acute ALD.
MATERIALS AND METHODS
Animals and treatments
Six- to eight-week-old male and female C57Bl/6J mice (C57 black 6 mice) (n = 5–9 per group) (Janvier S.A.S, Le-Genest-St-Isle, France) were housed in a pathogen-free barrier facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. All procedures were approved by the local Institutional Animal Care and Use Committee (V277/10EM, Stuttgart). Food and tap water were allowed ad libitum prior to experimentation and during alcohol exposure. Mice received either 6 g ethanol/kg body weight intragastically by gavage (needle size: 0.65 × 40 mm, WDT, Garbsen, Germany) or isocaloric (0.043 kcal/g body weight)/isovolumetric maltodextrin solution (control animals) in the morning. Twelve hours after the acute alcohol exposure animals were killed. In earlier studies and pilot experiments 12 h after the ingestion of a bolus dose of ethanol lipid accumulation in the liver and markers like PAI-1 and 4-HNE protein adducts were found to be markedly induced in both male and female mice (Bergheim et al., 2006a and unpublished data of our own group). This dose of ethanol does not cause mortality. Food intake was not assessed; however, during regular checkups on the animals (every 2 h) only small food intake was observed. Mice were sluggish, but conscious and regained normal behavior within ∼6 h of alcohol ingestion. Twelve hours after alcohol administration mice were anesthetized with 80 mg ketamine and 6 mg xylazin/kg body weight (i.p.). Portions of liver tissue were frozen immediately in liquid nitrogen and stored at −80°C for further analysis, while others were frozen-fixed in Tissue Tek® O.C.T.™ compound (Sakura Finetek Europe, Alphen aan den Rijn, Netherlands) for histological examination.
Ethanol concentration and activity of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in plasma
To determine blood alcohol levels the ‘alcohol dehydrogenase-enzymatic’ method described by Bonnichsen and Lundgren (Bonnichsen and Lundgren, 1957) was used (detection limit: 0.0125 µmol/µl). Levels of AST and ALT were determined using commercially available kits following the instructions of the manufacturer (both kits from Bioo Scientific, Austin, TX, USA). Alcohol levels as well as activity of AST and ALT were measured in heparinized plasma.
Hepatic lipid accumulation
Frozen sections of liver (10 μm) were stained with Oil Red O (Sigma, Steinheim, Germany) for 10 min and counter stained with hematoxylin (Sigma, Steinheim, Germany) for 45 s as described previously (Kanuri et al., 2009). Representative pictures of the Oil Red O staining at ×400 magnification were taken using a Zeiss microscope (Axio Vert 200M, Zeiss, Jena, Germany). Hepatic triglycerides were isolated and measured as previously detailed (Kanuri et al., 2009).
RNA isolation and real-time RT–PCR
For the detection of MyD88, IRF-3, PAI-1, Apolipoprotein B (ApoB), interleukin (IL)-6, the murine IL-8 homologue KC, monocyte chemotactic protein (MCP)-1 and 18 s mRNA transcript levels were quantified using an iCycler (Bio-Rad Laboratories, Munich, Germany) as described previously (Wagnerberger et al., 2012).
For the detection of hepcidin, ferritin, transferrin receptor (TfR) 1 relative mRNA transcript levels were quantified using the LightCycler FastStart DNA Master Hybridization Probes kit on a LightCycler (Roche, Mannheim, Germany) and applying the TaqMan methodology. The housekeeping gene β2-microglobulin (β2MG) was amplified in a parallel reaction for normalization. Primers were designed using the online Universal ProbeLibrary Assay Design Center (Roche, Mannheim, Germany) and synthesized at Eurofins MWG Operon (Ebersberg, Germany). Primer sequences are listed in Supplementary material, Table S1.
Immunostaining of 4-hydroxynonenal protein adducts and F4/80 positive cells in liver tissue
4-Hydroxynonenal (4-HNE) protein adducts staining and counting of F4/80 positive cells was performed as previously described (Bergheim et al., 2008; Wagnerberger et al., 2012) using a polyclonal primary antibody for 4-HNE (AG Scientific, San Diego, CA, USA) and a monoclonal primary antibody for F4/80 (Abcam, Cambridge, UK). For the detection of 4-HNE the extent of labeling in liver lobule was defined as percent of the field area within the default color range determined by the software using an image acquisition and analysis system incorporated in the microscope (×200; Axio Vert 200M, Zeiss, Jena, Germany). Numbers of F4/80 positive cells were counted at a magnification of ×400. For both stainings data from eight fields of each tissue section were used to determine means, respectively. All sections used for staining were prepared in parallel and sections for densitometric analyses were stained at the same time. Furthermore, analyses were performed simultaneously.
Occludin western blot
To extract duodenal protein snap-frozen samples of tissue obtained from the duodenum were homogenized in peqGOLD TriFast (Peqlab, Erlangen, Germany) and protein was isolated according to the manufactures instructions. Protein lysates (10–30 µg protein/well) were separated in a 10% SDS-polyacrylamide gel and transferred to Hybond™-P polyvinylidene difluoride membranes. The resulting blots were then probed with an antibody against occludin (Zymed, San Francisco, CA, USA) and bands were visualized using a Super Signal Western Dura kit (Pierce, Perbio Science, Rockford, IL, USA). To ensure equal loading, all blots were stained with Ponceau red. Signals of occludin were normalized to β-actin, which was detected using a commercially available antibody (New England Biolabs, Frankfurt, Germany). Protein bands were analyzed densitometrically using the Fluorchem Software (Alpha Innotech, San Leandro, CA, USA).
Microsomal triglyceride transfer protein activity
Measurement of microsomal triglyceride transfer protein (MTP) activity was performed with a commercially available kit according to the instructions of the manufacturer (Chylos, New York, NY, USA) as described previously (Kanuri et al., 2011).
Apolipoprotein B and TNFα enzyme-linked immunosorbent assay
Using commercially available enzyme-linked immunosorbent assay (ELISA) kits levels of ApoB and TNFα were determined in liver lysates following the instructions of the manufacturers (ApoB: Uscn Life Science, Inc., Huston, ID, USA; TNFα: Assaypro, St. Charles, IL, USA).
Statistical analyses
All results are given as mean ± SEM. Analysis of variances (ANOVA) with the post hoc test of Tukey was applied for the determination of significance levels (SPSS statistics, Version 17.0, 2008, SPSS, Inc., Chicago, IL, USA and Graph Pad Prism, Version 4.00, GraphPad Software, San Diego, CA, USA). Homogeneity of variances was tested using the Bartlett's test. A post hoc test was done when the means were significantly different in ANOVA. Grubbs' test was used to determine outliers. Differences were considered as significant if the P-value was <0.05.
RESULTS
Plasma alcohol levels and liver status 12 h after acute alcohol exposure
As to be expected by the results of Cope et al. (2000), small amounts of ethanol were detected in plasma obtained from controls regardless of sex, despite the fact that these animals were not challenged with ethanol; however, neither alcohol levels in plasma nor triglyceride and total lipid levels in livers differ among sexes (Figs 1 and 2). In contrast, plasma alcohol levels of female and male mice exposed to the ethanol solution were higher in both male (1.4-fold) and female (1.6-fold) mice compared with control animals (female mice: P < 0.05 in comparison with controls; male mice: n.s. in comparison with controls as levels varied considerably) (Fig. 1). Furthermore, in both male and female mice, acute ingestion of alcohol caused a significant accumulation of triglycerides and lipids in the liver in comparison with controls (Fig. 2A and B). However, despite similar alcohol doses per kg body weight and similar plasma alcohol levels, triglyceride levels and lipid accumulation were markedly more pronounced in livers of female mice than in those of male animals treated with ethanol (+ ∼3.3-fold, P < 0.05 in comparison with female alcohol-treated mice) (Fig. 2B). In both female and male mice, ethanol treatment was associated with an increase of ALT and AST levels in plasma (Table 1; ALT: female + ∼1.9-fold, male + ∼3.5-fold; AST: female + ∼2.7-fold, male + ∼1–4-fold).
Markers of liver injury, number of F4/80 positive cells and mRNA expression of pro-inflammatory cytokines in livers of female and male mice after acute alcohol treatmenta
| Female | Male | |||
|---|---|---|---|---|
| C | Ethanol | C | Ethanol | |
| ALT (U/l) | 7.0 ± 2.5 | 13.4 ± 1.7 | 6.5 ± 2.4 | 23.0 ± 2.8b,c,d |
| AST (U/l) | 31.1 ± 5.3 | 84.7 ± 5.4b,d | 47.4 ± 9.5 | 67.0 ± 9.3b |
| F4/80 positive cells (number per microscope field) | 18.8 ± 2.2 | 17.7 ± 0.6 | 19.9 ± 1.4 | 22.4 ± 2.7 |
| TNFα (ng/mg protein) | 0.15 ± 0.01 | 0.11 ± 0.01 | 0.07 ± 0.01b | 0.12 ± 0.02 |
| IL-6 mRNA expression (fold induction) | 9.4 ± 1.8 | 9.4 ± 1.9 | 8.5 ± 2.7 | 9.8 ± 4.7 |
| KC mRNA expression (fold induction) | 4.7 ± 2.2 | 4.7 ± 1.3 | 5.5 ± 1.3 | 2.3 ± 0.5 |
| MCP-1 mRNA expression (fold induction) | 3.2 ± 0.6 | 3.6 ± 0.6 | 2.2 ± 0.4 | 3.0 ± 0.6 |
| Female | Male | |||
|---|---|---|---|---|
| C | Ethanol | C | Ethanol | |
| ALT (U/l) | 7.0 ± 2.5 | 13.4 ± 1.7 | 6.5 ± 2.4 | 23.0 ± 2.8b,c,d |
| AST (U/l) | 31.1 ± 5.3 | 84.7 ± 5.4b,d | 47.4 ± 9.5 | 67.0 ± 9.3b |
| F4/80 positive cells (number per microscope field) | 18.8 ± 2.2 | 17.7 ± 0.6 | 19.9 ± 1.4 | 22.4 ± 2.7 |
| TNFα (ng/mg protein) | 0.15 ± 0.01 | 0.11 ± 0.01 | 0.07 ± 0.01b | 0.12 ± 0.02 |
| IL-6 mRNA expression (fold induction) | 9.4 ± 1.8 | 9.4 ± 1.9 | 8.5 ± 2.7 | 9.8 ± 4.7 |
| KC mRNA expression (fold induction) | 4.7 ± 2.2 | 4.7 ± 1.3 | 5.5 ± 1.3 | 2.3 ± 0.5 |
| MCP-1 mRNA expression (fold induction) | 3.2 ± 0.6 | 3.6 ± 0.6 | 2.2 ± 0.4 | 3.0 ± 0.6 |
aData are means ± SEM (female: n = 6–9; male: n = 5–6).
bP < 0.05 compared with female control animals.
cP < 0.05 compared with female alcohol-treated animals.
dP < 0.05 compared with male control animals.
Markers of liver injury, number of F4/80 positive cells and mRNA expression of pro-inflammatory cytokines in livers of female and male mice after acute alcohol treatmenta
| Female | Male | |||
|---|---|---|---|---|
| C | Ethanol | C | Ethanol | |
| ALT (U/l) | 7.0 ± 2.5 | 13.4 ± 1.7 | 6.5 ± 2.4 | 23.0 ± 2.8b,c,d |
| AST (U/l) | 31.1 ± 5.3 | 84.7 ± 5.4b,d | 47.4 ± 9.5 | 67.0 ± 9.3b |
| F4/80 positive cells (number per microscope field) | 18.8 ± 2.2 | 17.7 ± 0.6 | 19.9 ± 1.4 | 22.4 ± 2.7 |
| TNFα (ng/mg protein) | 0.15 ± 0.01 | 0.11 ± 0.01 | 0.07 ± 0.01b | 0.12 ± 0.02 |
| IL-6 mRNA expression (fold induction) | 9.4 ± 1.8 | 9.4 ± 1.9 | 8.5 ± 2.7 | 9.8 ± 4.7 |
| KC mRNA expression (fold induction) | 4.7 ± 2.2 | 4.7 ± 1.3 | 5.5 ± 1.3 | 2.3 ± 0.5 |
| MCP-1 mRNA expression (fold induction) | 3.2 ± 0.6 | 3.6 ± 0.6 | 2.2 ± 0.4 | 3.0 ± 0.6 |
| Female | Male | |||
|---|---|---|---|---|
| C | Ethanol | C | Ethanol | |
| ALT (U/l) | 7.0 ± 2.5 | 13.4 ± 1.7 | 6.5 ± 2.4 | 23.0 ± 2.8b,c,d |
| AST (U/l) | 31.1 ± 5.3 | 84.7 ± 5.4b,d | 47.4 ± 9.5 | 67.0 ± 9.3b |
| F4/80 positive cells (number per microscope field) | 18.8 ± 2.2 | 17.7 ± 0.6 | 19.9 ± 1.4 | 22.4 ± 2.7 |
| TNFα (ng/mg protein) | 0.15 ± 0.01 | 0.11 ± 0.01 | 0.07 ± 0.01b | 0.12 ± 0.02 |
| IL-6 mRNA expression (fold induction) | 9.4 ± 1.8 | 9.4 ± 1.9 | 8.5 ± 2.7 | 9.8 ± 4.7 |
| KC mRNA expression (fold induction) | 4.7 ± 2.2 | 4.7 ± 1.3 | 5.5 ± 1.3 | 2.3 ± 0.5 |
| MCP-1 mRNA expression (fold induction) | 3.2 ± 0.6 | 3.6 ± 0.6 | 2.2 ± 0.4 | 3.0 ± 0.6 |
aData are means ± SEM (female: n = 6–9; male: n = 5–6).
bP < 0.05 compared with female control animals.
cP < 0.05 compared with female alcohol-treated animals.
dP < 0.05 compared with male control animals.
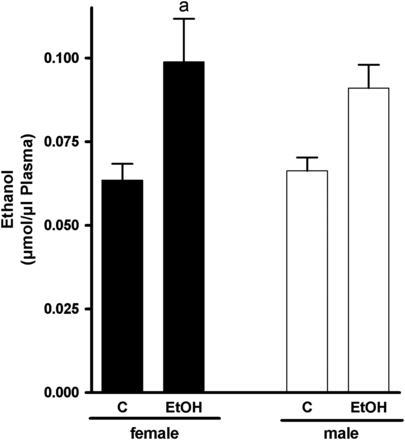
Effect of acute alcohol treatment on plasma alcohol concentration. Concentration of alcohol in plasma of female and male control mice and mice treated with alcohol 12 h after treatment. Data are shown as means ± SEM (female: n = 8–9, male: n = 5–6). aP < 0.05 vs. C female. C, control mice, EtOH, ethanol-treated mice (6 g EtOH/kg body weight).
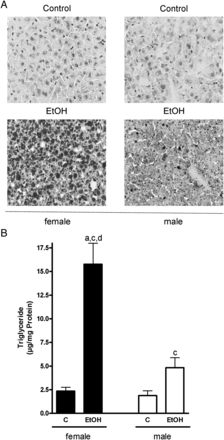
Effect of acute alcohol treatment on hepatic lipid accumulation. (A) Representative photomicrographs of the Oil Red O staining (×400) of livers of female and male control mice and mice treated with alcohol and (B) quantification of the hepatic triglyceride levels. Data are shown as means ± SEM (female: n = 9, male: n = 6). aP < 0.05 vs. female C, cP < 0.05 vs. male C, dP < 0.05 vs. male EtOH. C, control mice, EtOH, ethanol-treated mice (6 g EtOH/kg body weight).
Protein concentration of occludin in the duodenum and expression of MyD88 and IRF3 mRNA as well as concentration of 4-HNE protein adducts in the liver 12 h after acute alcohol exposure
As it has been shown before by others that acute and chronic ingestion of ethanol may alter intestinal microbiota and barrier function subsequently leading to an increase in systemic endotoxin levels (for overview see Bode and Bode (2005)) and as it recently has been shown that a decrease in the protein concentration of the tight junction protein occludin in the duodenum is associated with the etiology of other liver diseases associated with increased endotoxin levels (e.g. non-alcoholic fatty liver disease; (Spruss et al., 2012)), we determined protein levels of occludin in the duodenum. Protein levels of occludin in the duodenum did not differ between female and male mice regardless of additional treatments (Fig. 3E). Expression of the TLR-4 adaptor protein MyD88 in the liver did not differ between male mice treated with ethanol or controls, but was per se slightly lower than in female mice (n.s.). Despite no changes in the number of F4/80 positive cells in the liver (Table 1), in livers of female mice exposed to ethanol, expression of MyD88 was significantly induced by ∼2-fold in comparison with the respective controls (Fig. 3A). Similar results were found for IRF-3 mRNA expression (Fig. 3B); however, as expression levels varied considerable between animals, differences did not reach the level of significance. Protein levels of 4-HNE protein adducts were markedly lower in livers of male controls than in those of female controls (∼ −50%, P < 0.05). In livers of ethanol-treated male and female mice, levels of 4-HNE protein adducts were twice as high compared with their respective controls (Fig. 3C and D).
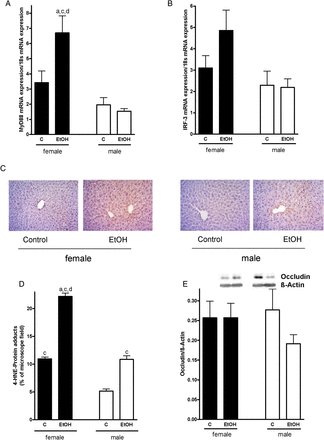
Effect of acute alcohol treatment on TLR-4 signaling cascade, hepatic lipid peroxidation and intestinal permeability. Relative hepatic mRNA expression of (A) MyD88, (B) IRF-3, both normalized to 18 s mRNA expression, (C) representative photomicrographs of immunostaining of 4-HNE protein adducts (×400), (D) densitometric analysis of 4-HNE adduct staining in liver sections and (E) densitometric analysis of occludin western blot in the duodenum of female and male control mice and mice treated with alcohol. Data are shown as means ± SEM (female: n = 6–9, C male: n = 4–6). aP < 0.05 vs. female C, cP < 0.05 vs. male C, dP < 0.05 vs. male EtOH. C, control mice, EtOH, ethanol-treated mice (6 g EtOH/kg body weight).
Markers of hepatic inflammation: TNFα protein levels and mRNA expression of IL-6, KC as well as MCP-1
To investigate whether markers of hepatic inflammation are also altered in a sex-specific manner after acute alcohol ingestion, we determined protein levels of TNFα and mRNA expression of IL-6, KC (murine IL-8 homologue) and MCP-1 in livers of male and female ethanol-treated mice. Protein levels of TNFα per se were lower in male control mice compared with female control animals; however, 12 h after ethanol treatment, no differences between control and ethanol-treated mice were found (Table 1). Neither sex nor alcohol ingestion had any effects on the mRNA expression of IL-6, KC and MCP-1 in livers of mice (Table 1).
Markers of iron metabolism in the liver 12 h after acute alcohol exposure
As iron overload in the liver is discussed to contribute to oxidative stress and to induce liver damage in experimental settings of treatment with alcohol (Bergheim et al., 2011), we determined markers of iron metabolism in livers of male and female alcohol-treated mice. Whereas hepcidin mRNA expression seemed not to be affected by the alcohol treatment, mRNA expression of hepcidin in the liver per se was significantly lower in male mice regardless of additional treatments in comparison with female animals. In contrast, mRNA expression of ferritin was significantly induced in livers of male and female alcohol-treated mice compared with their respective controls (Table 2). Expression of TfR1 mRNA in the liver was slightly higher in livers of mice exposed to alcohol in comparison with their respective controls; however, again, expression levels varied considerable among animals (Table 2).
Relative hepatic mRNA expression of hepcidin, ferritin and TfR1 in female and male mice after acute alcohol treatment, normalized to b2MGa
| Female | Male | |||
|---|---|---|---|---|
| C | Ethanol | C | Ethanol | |
| Hepcidin | 1.51 ± 0.17 | 1.29 ± 0.11 | 0.45 ± 0.08b,c | 0.29 ± 0.12b,c |
| Ferritin | 0.76 ± 0.08 | 1.16 ± 0.08b | 0.84 ± 0.06 | 1.45 ± 0.14b,d |
| TfR1 | 0.87 ± 0.10 | 1.01 ± 0.05 | 1.09 ± 0.12 | 1.20 ± 0.14 |
| Female | Male | |||
|---|---|---|---|---|
| C | Ethanol | C | Ethanol | |
| Hepcidin | 1.51 ± 0.17 | 1.29 ± 0.11 | 0.45 ± 0.08b,c | 0.29 ± 0.12b,c |
| Ferritin | 0.76 ± 0.08 | 1.16 ± 0.08b | 0.84 ± 0.06 | 1.45 ± 0.14b,d |
| TfR1 | 0.87 ± 0.10 | 1.01 ± 0.05 | 1.09 ± 0.12 | 1.20 ± 0.14 |
aData are means ± SEM (female: n = 9, male: n = 6).
bP < 0.05 compared with female control animals.
cP < 0.05 compared with female alcohol-treated animals.
dP < 0.05 compared with male control animals.
Relative hepatic mRNA expression of hepcidin, ferritin and TfR1 in female and male mice after acute alcohol treatment, normalized to b2MGa
| Female | Male | |||
|---|---|---|---|---|
| C | Ethanol | C | Ethanol | |
| Hepcidin | 1.51 ± 0.17 | 1.29 ± 0.11 | 0.45 ± 0.08b,c | 0.29 ± 0.12b,c |
| Ferritin | 0.76 ± 0.08 | 1.16 ± 0.08b | 0.84 ± 0.06 | 1.45 ± 0.14b,d |
| TfR1 | 0.87 ± 0.10 | 1.01 ± 0.05 | 1.09 ± 0.12 | 1.20 ± 0.14 |
| Female | Male | |||
|---|---|---|---|---|
| C | Ethanol | C | Ethanol | |
| Hepcidin | 1.51 ± 0.17 | 1.29 ± 0.11 | 0.45 ± 0.08b,c | 0.29 ± 0.12b,c |
| Ferritin | 0.76 ± 0.08 | 1.16 ± 0.08b | 0.84 ± 0.06 | 1.45 ± 0.14b,d |
| TfR1 | 0.87 ± 0.10 | 1.01 ± 0.05 | 1.09 ± 0.12 | 1.20 ± 0.14 |
aData are means ± SEM (female: n = 9, male: n = 6).
bP < 0.05 compared with female control animals.
cP < 0.05 compared with female alcohol-treated animals.
dP < 0.05 compared with male control animals.
Expression of PAI-1 mRNA and ApoB protein concentration as well as MTP activity in the liver 12 h after acute alcohol exposure
Acute ingestion of alcohol led to an ∼2-fold induction of PAI-1 mRNA expression in livers of female mice in comparison with their respective controls (n.s. as expression levels varied considerably; Fig. 4A). A similar effect was not found when determining PAI-1 mRNA expression levels in livers of male ethanol-treated mice; however, PAI-1 mRNA expression per se was slightly lower in livers of male mice compared with that found in livers of female animals (n.s.). As we had shown before that PAI-1 is involved in the modulation of hepatic triglyceride export (Kanuri et al., 2011) through mechanisms involving a modulation of the activity of the microsomal triglyceride transfer protein (MTP) we determined MTP activity and ApoB protein levels in the liver. In female mice, 12 h after the acute ethanol exposure, MTP activity was significantly higher than in the respective controls (+1.14-fold). In contrast, MTP activity did not differ between male ethanol-treated mice and controls (Fig. 4B). However, protein levels of ApoB were found to be significantly lower in livers of female mice exposed to ethanol in comparison with female controls. A similar effect on ApoB protein levels was not found in livers of male mice exposed to ethanol, but ApoB protein concentration per se was slightly lower in livers of male mice when compared with female animals (n.s.; Fig. 4C).
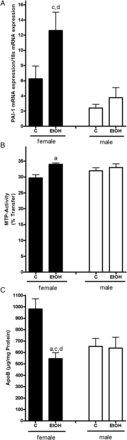
Effect of acute alcohol treatment on hepatic lipid export. (A) Relative hepatic mRNA expression of PAI-1, normalized to 18 s mRNA expression, (B) hepatic activity of MTP and (C) protein concentration of ApoB in livers of female and male control mice and mice treated with alcohol. Data are shown as means ± SEM (female: n = 6–8, C male: n = 4–6). aP < 0.05 vs. female C, cP < 0.05 vs. male C, dP < 0.05 vs. male EtOH. C, control mice, EtOH, ethanol-treated mice (6 g EtOH/kg body weight).
DISCUSSION
Acute alcohol-induced steatosis is more prominent in female than in male mice
Women have repeatedly been found to be more susceptible to ALD than men (Becker et al., 1996). A similar phenomenon has also been reported for chronic alcohol feeding models in rodents (Iimuro et al., 1997). However, whether gender-specific differences are also found after acute alcohol ingestion has, to our knowledge, not yet been systematically studied. In the present study, a mouse model of an acute alcohol binge consisting of one single dose of alcohol (6 g/kg body weight, given by gavage) was used to investigate whether female mice are also more susceptible to acute alcohol-induced hepatic steatosis than male animals. In the present study, a marked increase of AST and ALT plasma levels and accumulation of lipids and triglycerides was found in livers of both male and female mice 12 h after acute alcohol exposure; however, steatosis was markedly more prominent in livers of female than in male alcohol-treated mice. These results suggest that similar to the finding in chronic animal models of ALD and in humans with ALD, female gender is also in an acute setting of alcohol exposure associated with an increased susceptibility of the development of steatosis.
Results of previous studies in rodents further indicate that besides the shift in the ratio of NAD+ to NADH+H+, the markedly more pronounced liver damage observed after chronic intake of ethanol in the liver of female mice in comparison with male animals may be a result of an increased gut permeability (Bode et al., 1991; Parlesak et al., 2000), translocation of intestinal bacterial endotoxin (LPS) (Bode et al., 1987) and activation of TLR-4-dependent signaling cascades (e.g. MyD88- and/or IRF3-dependent). Indeed, it has been suggested by the results of Ikejima et al. (1998) that Kupffer cells of female rats react more sensitive when challenged with endotoxin than those of male rats. The activation of TLR-4-dependent signaling cascades in the liver has been shown to result in an induction of the formation of reactive oxygen species (ROS), release of TNFα and stimulation of hepatocytes (Enomoto et al., 1998; Ikejima et al., 1998) (for review also see Arteel (2003) and Bode and Bode (2005)). In the present study, despite no marked differences in duodenal protein levels of the tight junction protein occludin shown before in other models of liver damage (e.g. ob/ob mice and fructose-induced steatosis; (Haub et al., 2011; Wagnerberger et al., 2012)) to be indicative for an increased intestinal permeability, 12 h after the acute alcohol exposure, expression of both MyD88 and IRF-3 mRNA in the liver was markedly induced in female ethanol-treated mice, whereas a similar effect of the alcohol binge was not found in livers of male mice. However, contrary to the findings in settings of chronic alcohol exposure (Mandrekar et al., 2011), in the present study, other TLR-4-dependent pro-inflammatory markers like IL-6, KC (IL-8), MCP-1, TNFα or numbers of F4/80 positive cells were not altered in livers of male and female mice, respectively, when treated with ethanol. Recent studies in ovarectomized female rats that were treated with estrogen or testosterone and alcohol (2 weeks) have shown that alcohol consumption along with estrogen treatment increased liver inflammation by increasing NFκB translocation, TNFα mRNA and IL-6 protein (Lee et al., 2012). In contrast, anti-inflammatory effects of testosterone were due to an induction of IκB. In the present study, none of these parameters were altered. This might be due to the chosen model of alcohol ingestion (one single binge of ethanol vs. 2 weeks of ethanol feeding) and the time point (12 h after ethanol ingestion) of measurements. Further experiments will be needed to identify the potential role of estrogen in molecular mechanisms in the development of acute alcohol-induced liver steatosis. Taken together, the data of the present study indicate that in livers of female mice but not of male mice, TLR-4-dependent signaling cascades are at least in some parts still activated 12 h after an acute alcohol binge. The apparent discrepancy between occludin protein concentration and the induction of markers of an activation of the TLR-4-signaling cascade found in the present study may have resulted from the time point to detect levels of occludin in the present study. Indeed, results of Rivera et al. (1998) suggest that in female rats acute alcohol ingestion is associated with a marked increase in portal endotoxin levels as early as 60–120 min after alcohol exposure with a normalization of endotoxin levels thereafter. Therefore, it may be that in the present study, the time point to detect differences in tight junction protein levels was missed. However, it may also be that in settings of acute alcohol exposure other mechanism or the loss of other tight junctions may be involved in the increased intestinal permeability.
In line with the findings of others in models of chronic ALD (for overview see Arteel (2003)) levels of 4-HNE protein adducts, a marker of lipidperoxidation, were also found to be induced in livers of animals exposed to ethanol in the present study. Interestingly, contrary to the findings for MyD88 and IRF3, levels of 4-HNE protein adducts were markedly elevated in livers of both male and female mice exposed to ethanol. These data suggest either that in livers of male mice the TLR-4 signaling cascade was induced earlier and that this induction was no longer detectable 12 h after the acute alcohol binge or that other mechanisms may be involved. Results of animal experiments suggest that iron may prime Kupffer cells towards alcoholic liver disease (for overview see Xiong et al. (2003)). Indeed, it has been shown that an iron overload of the liver may contribute to oxidative stress and an activation of NFκB in Kupffer cells thereby contributing to the development of liver damage in experimental settings of ethanol-induced liver damage in rodents (for overview see Bergheim et al. (2011) and Xiong et al. (2003)). However, in line with the findings in patients with ALD but also rodent models (Suzuki et al., 2002; Ohtake et al., 2007; Jayaraman et al., 2012), in the present study, mRNA expression of ferritin and, to a lesser extent, of TfR1 was induced in livers of both male and female mice 12 h after the acute alcohol exposure suggesting that even one alcohol binge might be sufficient to alter hepatic iron deposition and/or metabolism. However, whether these alterations are sufficient also to prime hepatic Kupffer cells similar to what was found in settings of chronic alcohol exposure (Tsukamoto et al., 1999) remains to be determined. Somewhat contrary to the findings of other chronic models of ALD (Bridle et al., 2006; Harrison-Findik et al., 2006, 2007; Flanagan et al., 2007; Ohtake et al., 2007), mRNA expression of hepcidin, which was per se lower in livers of male mice, was not affected by the acute alcohol exposure. The differences between our results and those of others may be due to a shorter time of alcohol exposure, which might not be sufficient to suppress hepcidin mRNA expression in livers of alcohol-treated mice. Another reason might be that during acute alcohol exposure and consequent LPS-leakage from the gut, a part of the hepcidin-suppressive effect of ethanol is counterbalanced by hepcidin induction via the signal transducer and activator of transcription 3, the inflammatory signaling pathway responsible for hepcidin induction during inflammation (Verga Falzacappa et al., 2007). However, this will have to be addressed in future studies. Taken together, the results of the present study suggest that similar to the findings in models of chronic alcohol-induced liver damage, the acute ingestion of large amounts of alcohol and associated lipid accumulation may result in an induction of the TLR-4 signaling cascades, formation of ROS and some markers of iron metabolism in the liver in female mice. However, our results also suggest that the activation of the TLR-4-dependent signaling pathways in livers of male mice after acute alcohol ingestion was either less pronounced (e.g. only at the level of already present protein vs. at the level of transcription), faster terminated or is not causally involved in the lipid accumulation. This will have to be addressed in future studies.
It has further been shown that the increased release of TNFα in models of acute and chronic alcohol exposure is associated with an induction of PAI-1, which through HGF/c-Met-dependent pathways leads to an impairment of the hepatic MTP-mediated triglyceride export via VLDL (Sugimoto et al., 2002; Tomita et al., 2004; Bergheim et al., 2006b). However, whether alterations in triglyceride export are also regulated sex-specifically under acute alcohol exposure has not yet been studied to our knowledge. In the present study, acute alcohol ingestion resulted in a significant induction of PAI-1 mRNA expression but also of MTP activity in livers of female mice but not in male animals. In contrast, ApoB protein levels were markedly lower in livers of female mice exposed to alcohol, whereas in male mice a similar effect of the alcohol ingestion was not found. Results of chronic ethanol-feeding experiments (37 days) indicate a decreased hepatic MTP activity as well as mRNA expression in male Sprague–Dawley rats (Sugimoto et al., 2002). Results of other studies using a model of acute ethanol ingestion (1 or 3 g/kg) in male Sprague–Dawley rats also suggest a decrease of hepatic MTP mRNA expression 3 h after ethanol administration (Lin et al., 1997). In the present study, the ‘discrepancy’ between PAI-1 induction and MTP activity might be due to the chosen time point of 12 h. It could be that MTP activity may be reduced at an earlier time point and that MTP activity already increases again 12 h after ethanol ingestion. Taken together, our data suggest that the sex-specific differences in the susceptibility towards acute alcohol-induced liver steatosis may at least partly be due to an induction of PAI-1 and a loss of ApoB protein in livers of female mice not found in livers of male mice. However, these data by no means preclude that other mechanisms may also be involved in the higher susceptibility of female mice towards acute alcohol-induced liver damage.
In conclusion, our data suggest that similar to the findings in humans and animal models of chronic alcohol exposure, female mice are also more susceptible to acute alcohol-induced fatty liver than male mice. Our data further suggest that the higher susceptibility of female mice to acute alcohol-induced liver steatosis is associated with an increased activation of TLR-4-dependent signaling cascades and up-regulation of PAI-1 in livers of female mice. However, future studies will have to address not only the underlying molecular mechanisms but also whether these mechanisms are associated with the higher susceptibility to ALD in women.
Supplementary material
Supplementary material is available at Alcohol and Alcoholism online.
Funding
The work was supported by a grant from Danone Institute Nutrition for Health, Germany, to I.B.
Conflict of interest statement. None declared.
Acknowledgments
The authors thank Ms Teresa Peccerella, Heidelberg, for technical assistance.

{kind=link}
{kind=link}
{kind=link}
{kind=link}