Article Text

Abstract
Introduction Dietary patterns that might induce remission in patients with active Crohn’s disease (CD) are of interest to patients, but studies are limited in the published literature. We aim to explore the efficacy of the CD therapeutic dietary intervention (CD-TDI), a novel dietary approach developed from best practices and current evidence, to induce clinical and biomarker remission in adult patients with active CD.
Methods and analysis This study is a 13-week, multicentre, randomised controlled trial in patients with mild-to-moderate active CD at baseline. One hundred and two patients will be block randomised, by sex, 2:1 to the intervention (CD-TDI) or conventional management. Coprimary outcomes are clinical and biomarker remission, defined as a Harvey Bradshaw Index of <5 and a faecal calprotectin of <250 µg/g, respectively.
Secondary outcomes include gut microbiota diversity and composition, faecal short-chain fatty acids, regulatory macrophage function, serum and faecal metabolomics, C reactive protein, peripheral blood mononuclear cell gene expression profiles, quality of life, sedentary time and physical activity at 7 and/or 13 weeks. Predictive models of clinical response to a CD-TDI will be investigated.
Ethics and dissemination The research protocol was approved by the Conjoint Health Research Ethics Board at the University of Calgary (REB19-0402) and the Health Research Ethics Board—Biomedical Panel at the University of Alberta (Pro00090772). Study findings will be presented at national and international conferences, submitted for publication in abstracts and manuscripts, shared on social media and disseminated through patient-education materials.
Trial registration number NCT04596566.
- Crohn's disease
- inflammatory bowel disease
- diet
- clinical trials
Data availability statement
Data sharing not applicable as no datasets were generated and/or analysed for this study.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Canada and the USA have among the highest rates of inflammatory bowel disease (IBD) in the world, with 1 in 360 and 1 in 503 people affected, respectively.1 2 Importantly, up to 30% of patients with IBD do not respond to induction therapy with biologics and over 40% of patients will eventually lose response to pharmacotherapy.3 The response rates to subsequent biologic therapies are diminished with each treatment failure, increasing the risk for surgery and reducing health-related quality of life (HRQoL), resulting in increased healthcare costs.4 Furthermore, IBD-related treatments can have significant adverse consequences, including an increased risk of serious infections and malignancies. Potential treatment failure and the risk of adverse consequences to medication highlight the importance of identifying new treatments to alter the disease course of IBD.
The topic of diet and its use as primary therapy to manage intestinal inflammation is a high-priority area of interest for patients with IBD and the role of precision nutrition and its application is of emerging interest. The precision nutrition concept postulates that the ‘one diet fits all’ approach is suboptimal and that tailoring diets based on predictive criteria can optimise health outcomes for a given individual. Yet, there is a limited, although growing, body of high-quality evidence in support of comprehensive dietary recommendations as therapy for IBD.
Research suggests that IBD arises from an interaction between genetic and environmental influences5 but the pathogenesis of IBD is not completely understood. Observational studies have examined associations between diet and the development of IBD6 and retrospective studies have demonstrated a higher incidence of Crohn’s disease (CD) in patients who consume a western dietary pattern.6–8 Diet plays a key role in defining the composition of the human gut microbiota and, consequently, the production of microbial metabolites.9 A disruption in the composition of the gut microbiota and metabolites can result in the development of intestinal inflammation.10 Moreover, the symbiotic relationship between the gut microbiota and the host contributes to epithelial barrier function, which is critical to the maintenance of intestinal immune homeostasis; disturbance of the epithelial barrier enhances inflammatory responses via activation of the mucosal immune system and is believed to contribute to the risk of development of IBD.11 Despite these proposed mechanisms for dietary relationships to gut inflammation, there are few published reports about which foods or dietary patterns may promote induction of disease remission in patients with active inflammation. Yet, patients with IBD frequently have a strong belief that diet plays a key role in controlling the course of their disease and may be a trigger of disease relapse.2 12–14
Recent studies have demonstrated a relationship between diet and disease remission. The landmark Crohn’s Disease Exclusion Diet (CDED) Study in 74 children with mild-to-moderate CD showed higher sustained remission rates, defined as a Pediatric CD Activity Index (PCDAI) Score below 10, in those who received 12 weeks of the intervention diet compared with those who received exclusive enteral nutrition with reintroduction of habitual diet, a gold-standard therapy in paediatric patients with CD.15 In another recent randomised controlled trial (RCT) that compared the effectiveness of the specific carbohydrate diet (SCD) with the Mediterranean diet (MD) for 12 weeks as a treatment for CD in adult patients with mild-to-moderate symptoms, both diets induced symptomatic remission in approximately 45% of patients.16 Among patients with elevated baseline faecal calprotectin (FCP), a validated biomarker of intestinal inflammation, 34.8% (8/23) and 30.8% (4/13) of patients in the SCD and MD groups, respectively, achieved biomarker remission. However, that RCT did not include a placebo arm to compare the effectiveness of the diets. In a separate proof-of-concept trial of paediatric patients with active CD, a dietary intervention using whole foods to replicate the gut microbiome that is observed with the use of an enteral nutrition formula induced a clinical response, defined as a weighted PCDAI (wPCDAI) Score decrease≥17.5, in 80% of patients and remission, defined as a wPCDAI Score<12.5, in 60% of patients after 8 weeks.17 Lastly, in a pilot RCT in adults with ulcerative colitis (UC) with active disease, the efficacy of a novel UC diet alone compared with the UC diet with faecal transplant or faecal transplant alone was explored.18 Disease activity was defined by a Simple Colitis Activity Index≥5 but <11 and an endoscopic Mayo Score of 2–3, refractory to medications. The UC diet alone appeared to promote clinical remission (6/15=40%) better than faecal transplant with (4/19=21.1%) or without (2/17=11.8%) the UC diet, although this did not reach statistical significance. However, mucosal healing was achieved more frequently in patients receiving the UC diet alone (3/15=20%, p<0.05) than in patients who received a faecal transplant with (0/19) or without (0/17) the UC diet.
Despite this evidence that diet may influence disease remission, there is still a lack of well-defined relationships between dietary determinants and disease relapse. Apart from changes in the gut microbiota that were observed with the CDED, other mechanisms of action to explain clinical and biomarker response or predictive characteristics to identify diet respondents were not determined in these past studies. Interindividual differences arising from gut microbiome composition, genetics, lipidomics, immune function and other factors, such as physical activity, may explain the heterogeneous response to dietary therapies. Identifying clinical and biosignature responses to dietary interventions in patients with CD through recent advances in microbial analysis and other ‘omic’ technologies that integrate machine learning principles to understand large data sets could inform on the effects of diet on various proposed mechanisms of disease-related inflammation, which could allow the development of personalised dietary recommendations.
Given advancements in our understanding of dietary mechanisms of action in CD, we recently developed the CD Therapeutic Dietary Intervention (CD-TDI), a state-of-the-art, evidence-based dietary approach that incorporates global principles of the MD refined to inform specific food choices based on the results from our pilot data and from published literature reporting mechanisms of mitigating inflammation in IBD.15 17 19 20 The CD-TDI is a whole-food diet focused on adequate intakes of green leafy vegetables and other foods rich in beta-carotene, soluble fibre and resistant starch and optimal intakes of flavanols. It promotes an optimal n-6:n-3 poly-unsaturated fatty acid ratio (8:1) associated with C reactive protein (CRP) levels≤5 mg/L,20 together with therapeutic proportions of monounsaturated fatty acids and recommends abstinence from processed foods and food additives.21–23
In the present multicentre RCT, we aim to explore the efficacy of the CD-TDI compared with conventional management (CM) to induce clinical and biochemical remission in symptomatic and asymptomatic patients with active, mild-to-moderate luminal CD. We also aim to develop predictive models of clinical response to a CD-TDI using host biological pathways, microbiome metagenomics, metabolomics, nutrigenomics and activity levels.
The specific objectives of this study are to:
Compare the proportion of patients in each study group at week 13 who are in clinical remission, defined as a Harvey Bradshaw Index (HBI) of <5.
Compare the proportion of patients in each study group at week 13 who are in biochemical remission, defined as an FCP of <250 µg/g.
Examine whether the CD-TDI had a significant effect on key clinical, laboratory, patient-reported and behavioural outcomes, including gut microbiota diversity and composition, faecal short-chain fatty acids (SCFA), regulatory macrophage function (ie, phagocytosis and cytokine output, impact on epithelial wound healing and barrier function), red blood cell fatty acid profiles, serum and faecal metabolomics, nutrigenomics, serum CRP, peripheral blood mononuclear cell (PBMC) gene expression, quality of life, physical activity and sedentary time, at or before 13 weeks, and to assess whether these parameters or changes in parameters predict response to the diet intervention.
Methods and analysis
Study design
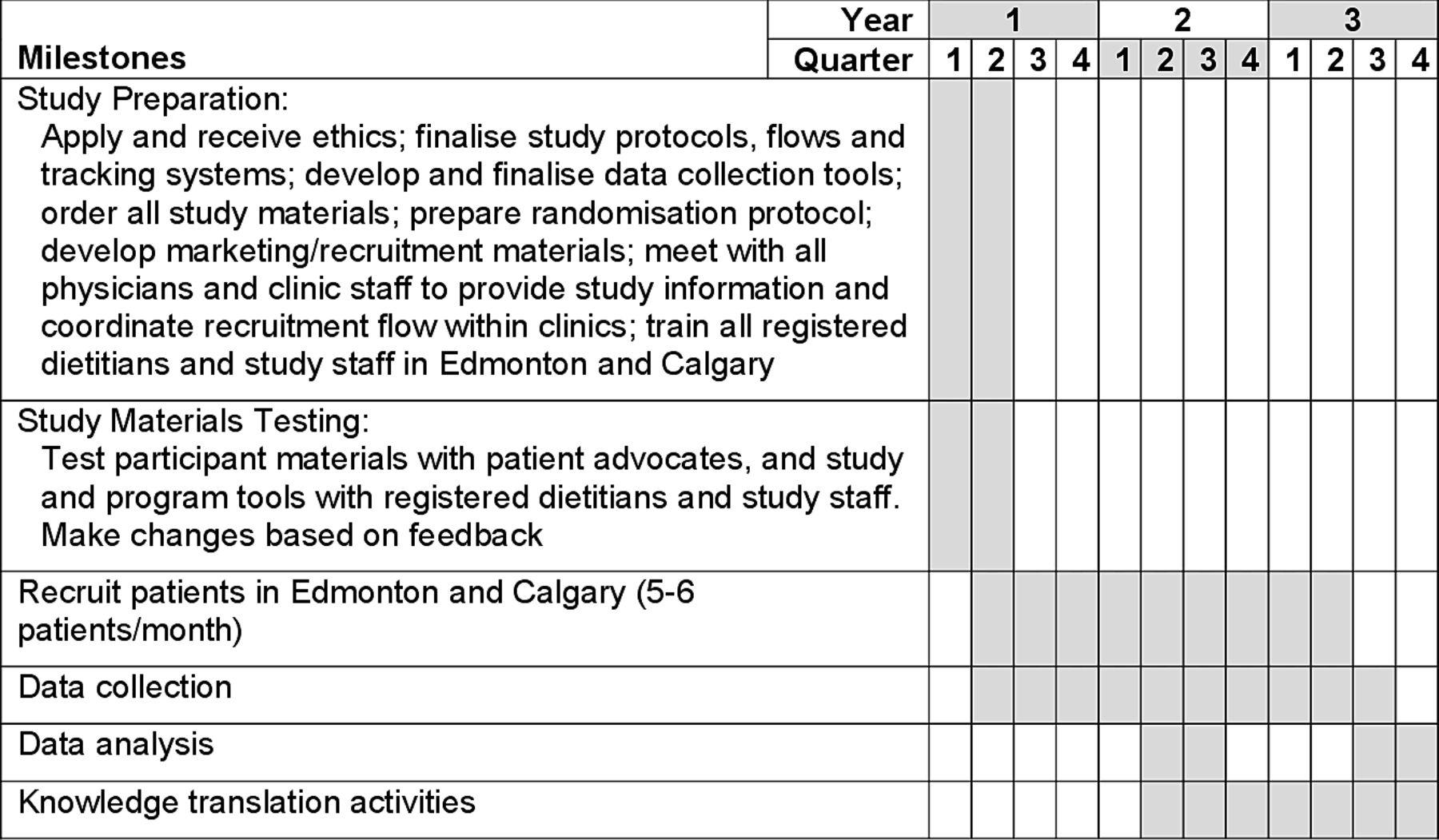
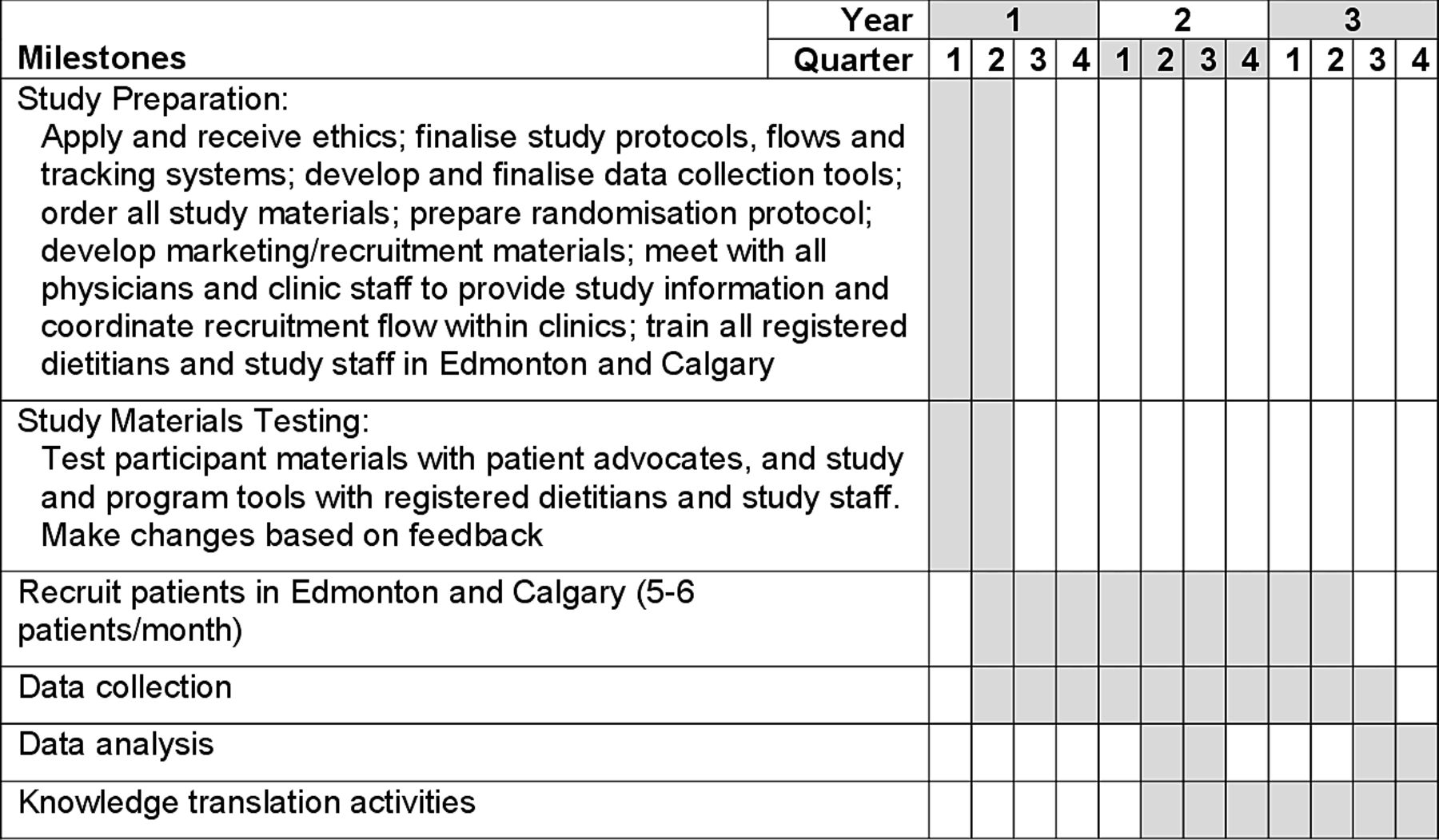
This study is a 13-week, investigator-blinded, multicentre RCT conducted at the University of Calgary (Calgary, Alberta, Canada) and University of Alberta (Edmonton, Alberta, Canada). Eligible participants will be randomly assigned to either the intervention group (CD-TDI) or CM control group. Healthy volunteers will be recruited to provide blood samples for the macrophage analysis only. See figure 1 for a schematic of the study design and figure 2 for study timelines.
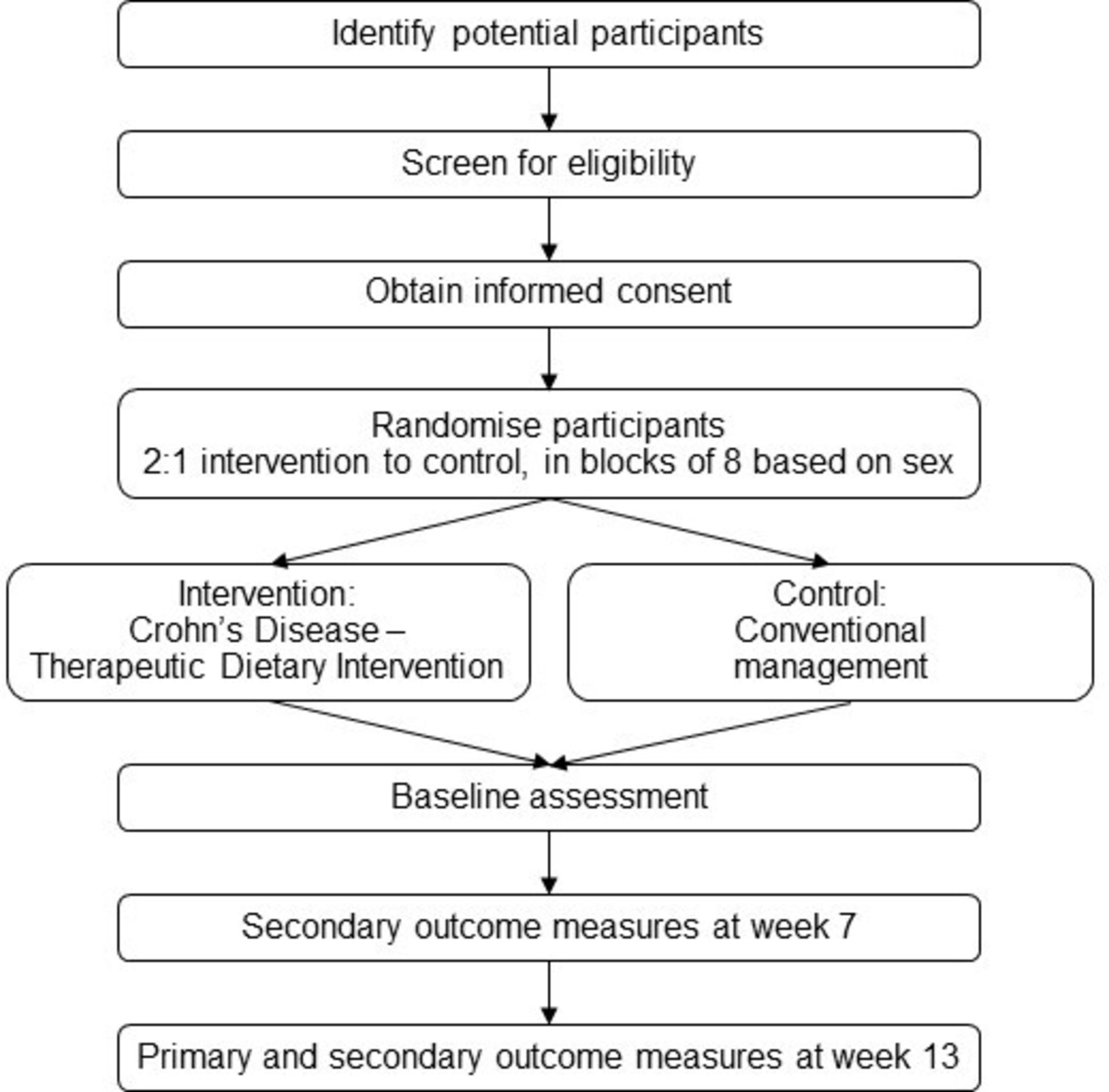
Study design.
{kind=link}
{kind=link}
Study timelines.
Participants
Study participants will be symptomatic with mild-to-moderate CD (defined as an HBI>5 and<16) or asymptomatic (defined as an HBI<5) with an elevated FCP (≥250 µg/g) at the time of recruitment. The trial’s specific inclusion and exclusion criteria are listed below.
Inclusion criteria
Adult patients≥18 years of age.
Clinical diagnosis of luminal ileal, ileo-colonic or colonic CD.
HBI<16 at the time of recruitment.
Biomarker evidence of inflammation at enrolment, defined as an FCP≥250 µg/g within 4–6 weeks prior to recruitment.
Oral prednisone not prescribed or prescribed up to a maximum of 30 mg/day for 2 weeks, with a fixed taper of 5 mg/week.
Stable biologic, immunosuppressant or aminosalicylate dosing for 4 weeks prior to study enrolment.
Able to provide informed consent.
The potential effects of baseline oral prednisone use on measures taken at week 7 will be controlled for as follows: (a) all participants prescribed the steroid will follow the same tapering regimen, (b) the randomisation will be stratified by steroid use, (c) the number of patients using the steroid should be equal based on the randomisation and (d) results can be analysed by steroid usage to ensure the effect is the same. Not allowing steroid exposure would exclude many symptomatic patients, making it difficult to recruit enough participants to meet the required sample size and limiting real-world application.
Exclusion criteria
HBI≥16 at the time of recruitment.
Upper gastrointestinal tract CD.
Evidence of active perianal or fistulising disease.
>One small bowel resection.
Significant chronic disorders such as cardiac disease, renal failure and active pulmonary disease.
Any psychiatric or neurocognitive disorder that would limit ability to participate.
Use of a laxative within 1 month prior to recruitment.
Use of antibiotics within 2 months prior to recruitment.
Presence of an ostomy.
Any change (ie, starting, stopping or changing dose) in medication for CD, apart from corticosteroids, within 4 weeks prior to recruitment.
COVID-19 within 8 weeks prior to recruitment or during the study.
Pregnant or breast feeding.
Recruitment
Multiple methods will be used to secure participants for this study. First, participants will be recruited from two world-class IBD centres in Alberta, Canada, that receive >100 referrals per month at each site for routine clinical assessment of patients with IBD. The treating IBD gastroenterologist will discuss the study with eligible patients and will formally refer patients who are agreeable to the study. Second, the study will be advertised through patient foundations, such as the Canadian Digestive Health Foundation and Crohn’s and Colitis Canada. Finally, the study will be promoted using social and print media. If patients self-refer, the study team will review their eligibility criteria prior to enrolling them.
Randomisation and blinding
Patients will be screened for eligibility and will provide their informed consent to participate. They will be randomly assigned in a 2:1 ratio to the CD-TDI group or the CM group, in blocks of eight based on sex. Randomisation will be done using a computerised random number generator and the group assignment will be concealed from the study coordinator enrolling patients, using sealed envelopes, until the time of assignment. All physicians, including treating physicians and study investigators, who are completing clinical primary outcome measures will be blinded to the study group.
Intervention: CD therapeutic diet intervention
Patients in the CD-TDI group will meet virtually with a registered dietitian (RD) trained in the CD-TDI protocol every week for 13 weeks of patient-centred counselling. The goals of the counselling are to promote adherence to the CD-TDI and to assess for and treat malnutrition, including both dietary macronutrient and micronutrient deficiencies, using whole foods.
The counselling protocol will incorporate behaviour change techniques that have been shown to promote increased dietary self-efficacy, such as self-monitoring, goal setting with review, contingent rewards, operant conditioning and revising negative thinking strategies.24 Programme materials will include structured 1-week meal plans; recipes; lists of foods that are mandatory, allowed or to be avoided; specific food goals and face-to-face counselling delivered virtually. Patients will prepare their own foods with guidance from the RD to meet the requirements of the CD-TDI while making acceptable adjustments to suit their preferences and nutritional status. For example, the RD would counsel a patient who was diagnosed with malnutrition to optimise protein and energy intake while retaining the principles of the CD-TDI. Patient compliance will be measured weekly using diet adherence checklists that will be completed during the virtual visits with the RD. The RD will assess which food goals are being met, and these goals will then be translated into an adherence score.
Control: CM
Patients receiving CM will meet virtually with the same RD as patients in the CD-TDI group at baseline and week 13 to complete a dietary assessment at each timepoint. They will be advised to follow their habitual diet during the study. On completion of the study, patients in the CM group who are still experiencing a disease flare will be offered the CD-TDI if they are interested.
Safety and adverse events
Participants will be screened and monitored for malnutrition. The study RD will assess participants for malnutrition using the abridged patient-generated subjective global assessment, a validated tool to determine malnutrition status in patients with chronic disease,25 at weeks 0, 7 and 13 for patients in the CD-TDI group and at weeks 0 and 13 for patients in the control group. Patients who are malnourished will continue to participate in the study. Those in the CD-TDI will receive counselling from the study RD, while those in the control group will be referred to the IBD clinic RD. Any participants who continue to be malnourished at the end of the study will be referred to the IBD clinic RD for further care.
Participants will be screened at baseline for anxiety. The RD or study coordinator will score the patient’s anxiety using the generalised anxiety disorder (GAD)-7 scale, a validated seven-item questionnaire commonly used to measure anxiety risk.26 Patients who score at risk of moderate or severe anxiety will be offered referral to the IBD clinic psychologist and will continue to participate in the study.
The RD or study coordinator will ask participants about adverse events, such as weight loss, malnutrition, hospitalisation and infection, weekly during virtual follow-up visits and record reported events on the designated form.
Data collection
Baseline characteristics
Demographic (sex, age, smoking status, body mass index, medications) and clinical (HBI, Montreal classification of disease behaviour, number of courses of systemic corticosteroids and hospitalisations for CD in the past 2 years) data will be captured at baseline.
Dietary assessment
All participants will complete two non-consecutive 24 hours dietary recalls with an RD at baseline (week 0) and after 13 weeks using the five-step multiple-pass method, a structured interview to capture detailed information about all the foods, beverages and supplements consumed in the past 24 hours.27 28 Details about oral nutrient supplements, probiotics and non-steroidal anti-inflammatory drugs will be captured at weeks 0 and 13 for both groups and during weekly visits with the RD for participants in receiving the CD-TDI.
Coprimary outcomes
Coprimary outcomes will be assessed at week 13. They are: (1) symptomatic clinical remission defined by an HBI<5 and (2) objective biochemical remission defined by an FCP<250 µg/g. HBI and FCP will also be assessed as secondary outcomes at week 7. Patients who require dose intensification or a change in biologic after the start of the study will be considered treatment failures.
Harvey Bradshaw Index
The HBI is a validated, non-invasive clinical measure of disease activity used to determine CD clinical disease severity.29 An HBI<5 will indicate clinical remission. The referring gastroenterologist will complete the HBI at baseline and study investigators will complete the HBI at weeks 7 and 13.
Faecal calprotectin
The FCP test is used to detect inflammation in the colon and is associated with disease activity and severity.30 FCP levels will be assessed at weeks 0, 7 and 13. FCP levels will be determined by quantitative ELISA (Bühlmann fCAL ELISA, EK-CAL) according to the manufacturer’s instructions.
Secondary outcomes
The effect of the CD-TDI on a number of secondary outcomes to inform mechanisms of action and the ability of these outcomes to predict response to the CD-TDI will be assessed. Given the relationship between the gut microbiota, bacterial metabolites, immune cells, PBMCs, monocytes, lipid subtypes and intestinal inflammation, changes in these measures will be evaluated. CRP as a secondary outcome will be measured as a marker for CD activity, with higher levels of CRP associated with more severe disease activity.31 Changes in HRQoL and sedentary time will be assessed as the key patient-reported and behavioural outcomes, respectively.
Faecal microbiome sequencing
Taxonomic and functional gut microbiome composition will be measured using agnostic shotgun metagenomic sequencing. Microbiome sequences will be analysed for alpha diversity, beta diversity, species and functional composition and biomarker discovery. The gut microbiome composition will be calculated for each participant at weeks 0, 7 and 13.
Serum and faecal metabolome
Small polar metabolites will be extracted from serum and faecal samples using ice-cold methanol. Samples will be homogenised in a TissueLyser and centrifuged and supernatants will be stored at −80°C until analysis. Hydrophilic interaction liquid chromatography with mass spectrometry (HILIC–MS/MS) will be performed as previously described32 using a Q Exactive HF Hybrid Quadrupole-Orbitrap Mass Spectrometer coupled to a Vanquish UHPLC System. Concentrations of faecal SCFAs will be measured according to our previously published protocol.33 Metabolites will be identified using retention times and MS profiles of a library of metabolite reference standards. All data will be analysed using MAVEN and metabolite levels will be reported as relative intensities (untargeted analyses) or as concentration (targeted analyses of SCFA). Serum and faecal metabolites will be assessed at weeks 0 and 13 and SCFAs will be measured at weeks 0, 7 and 13.
Macrophages
PBMCs will be collected and cultured (4 hours, 37°C) on Petri dishes in RPMI-1640 medium with 2% penicillin/streptomycin, Hepes (20 mM) and 10% pooled normal human heat-inactivated, sterile-filtered AB serum (Innovative Research, Novi, Michigan).34 Non-adherent cells will be removed and adherent monocytes collected and resuspended at 1×106 cells/mL and incubated in human macrophage-colony stimulating factor (hM-CSF; 10 ng/mL). Culture medium will be changed on day 5 and hM-CSF will be added. The resultant macrophages will be unstimulated (M(0)) or treated with human recombinant interferon (IFN)-γ (M(IFNγ)) or interleukin (IL)−4 (M(IL4)) (both 20 ng/mL, 48 hours, 37°C) for conversion to classically activated (CAM) and alternatively activated macrophages (AAM), respectively. Successful conversion to CAM or AAM will be determined by quantitative PCR of marker genes: increased CD206 and CCL18, reduced CD14 mRNA expression in M(IL4)s and increased CNDP1 mRNA in M(IFNγ). The function of M(0), M(IFNγ) and M(IL4) will be assessed as follows: (1) phagocytosis of inert fluorescent latex beads and green fluorescent protein-expressing Escherichia coli;35 (2) conditioned medium (CM) collected and applied to monolayers of the human colon-derived T84 epithelial cell line that have received a sterile wound with a razorblade and epithelial migration (a surrogate for wound healing) assessed 24 hours later;34 and (3) electrically confluent (ie, transepithelial electrical resistance (TER)>800 Ω cm2) filter-grown monolayers of T84 cells treated with macrophage CM and barrier assessed by change in TER and transepithelial fluxes of 4 and 70 kDa fluorescein isothiocyanate-dextran (Sigma Chemical Co.).36 Macrophage analysis will be conducted at weeks 0 and 13.
Nutrigenomics
PBMCs and monocytes will be isolated from peripheral blood before and after the CD-TDI via standard procedure and RNA will be extracted using commercial kits. The quality and quantity of total RNA will be assessed using an Agilent Bioanalyzer. Next, total RNA will be hybridised to Affymetrix Human 2.1 ST Array Strips to investigate 28 000 coding transcripts. Microarray data will be tested for quality control and normalised using tacrolimus. Nutrigenomics and transcriptomics will be performed at weeks 0 and 13.
Lipidomics
Fatty acid profiles will be determined by analysing red blood cell membrane phospholipids and faecal samples for excreted fatty acids using flame ionisation detector gas chromatogram using standards specific to medium-chain and long-chain fatty acids, similarly described.37 Lipidomics will be performed at weeks 0 and 13.
Serum biochemistry
CRP will be measured at weeks 0 and 13, along with other laboratory measures, including albumin, prealbumin, complete blood count, electrolytes, creatine, magnesium, phosphate, folate, zinc, ferritin and vitamins A, B12 and D (25OHD3).
Health-related quality of life
HRQoL as a patient-reported outcome will be assessed using the 12-item short-form health survey V.1 (SF-12)38 at weeks 0, 7 and 13. The SF-12 measures eight health domains to provide physical and mental health composite scores.
Physical activity and sedentary time
The main behavioural outcome in this trial is sedentary time, which will be assessed by using the activPAL inclinometer (PAL Technologies, Glasgow). Inclinometers are small electronic devices worn discretely on the upper thigh. They measure duration and frequency of time spent sitting, standing and stepping (light ambulation) and number of postural changes. ActivPAL monitors are the most validated and widely used devices for measuring sitting, standing and moving/stepping (and transitions in between). Physical activity (ie, light, moderate and vigorous intensity activity) will be assessed using the Actigraph GT3X+ accelerometer (Actigraph, Pensacola, Florida). The GT3X+ records acceleration/motion using a triaxial accelerometer and is worn on an elasticised belt on the waist, over one hip, during waking hours for 7 consecutive days at each data collection time point. Participants will fill in an activity log each day to record the time the waist-worn accelerometers are put on and taken off each day, and the time the patient goes to sleep and wakes up. The GT3X+ provides second-by-second data on sedentary time (ie, sitting/lying/standing), steps and all intensities of movement including light, moderate and vigorous intensity activity.
Sample size
The sample size calculation is based on the primary outcome of symptomatic (clinical) and objective (biochemical) remission. We estimated the effect size estimates in the control group based on corticosteroid-free remission rates in the phase III CALM trial, wherein 23.8% (29/122) of patients in the conventional therapy arm achieved remission at week 11.39 Assuming a control group remission rate of 25%, an alpha level of 0.05, power of 0.80 and 2:1 allocation ratio (CD-TDI to CM), a sample size of 93 patients (62 and 31 patients randomised to CD-TDI and CM, respectively) would be sufficient to detect a remission rate 55% in the intervention arm. Estimates of the remission rate in the CD-TDI arm are based on a previous prospective trial of a similar dietary intervention in paediatric patients with mild-to-moderate CD.15 In this trial, 76% (28/37) of patients achieved clinical remission at week 12. Given that adding biochemical measures is likely to reduce the remission rate compared with symptoms alone and because paediatric patients may be more responsive to dietary interventions compared with adults, we used a more conservative estimate of remission in the CD-TDI arm of 55% for the power calculations. Assuming a 10% drop out rate, an estimated 102 patients must be randomised.
Data analysis
Demographic and clinical characteristics of the study groups will be assessed using descriptive statistics. Statistical significance will be tested at an alpha=0.05, two-sided level of significance. For the primary efficacy analysis, the proportion of patients in each study group who have achieved the coprimary endpoints of clinical and biomarker remission will be compared using the χ2 test. Several sensitivity analyses are planned, including: (1) evaluating the proportion of remitters among initially symptomatic versus asymptomatic patients; (2) evaluating the proportion of patients who achieve clinical and biomarker endpoints separately and (3) using last-observation-carried-forward imputation for patients with missing data.40 As a conservative default, non-responder imputation will be implemented but the pattern-mixture model will be used41 to assess the potential impact of missing data on study conclusions.42 Generalised linear mixed models will be used to assess the effect of additional covariables that may confound or interact with treatment groups, and the presence of effect modification by sex, IBD disease phenotype and disease activity at baseline will be evaluated by testing for significance of the interaction term. Intention-to-treat analysis will be done for all analyses by including all patients within their group assignment.
Faecal calprotectin
FCP levels will be analysed in duplicate and quantify them using a standard curve (30–1800 µg/g) from calibrators included in the ELISA kit.
Faecal microbiome
For microbiome sequencing data quality, control of raw FASTQ files will be performed prior to machine learning analysis using software tools such Trimmomatic43 and BMTagger44 to obtain high-quality non-host reads. Reads of sufficient quality will be mapped using MetaPhlAn345 and HUMAnN345 to obtain high-resolution taxonomic and functional abundance profiles that include relative abundance and/or read counts. Unsupervised learning strategy based on robust principal component analysis will be used to explore the major variability in the data and to detect outliers. Kyoto Encyclopedia of Genes and Genomes annotations and functional diversity profiling will also be performed and web-based tools will be used to assign metagenomic results into different functional groups.
Serum and faecal metabolome
Metabolites will be identified based on exact mass, MS/MS fragmentation patterns and coretention of metabolites with analytical reference standards. Data will be peak picked using MAVEN and data will be analysed using in-house software tools developed in R statistical software package. SCFA concentrations will be determined by the 12C/13C signal intensity ratio and the respective 13C-internal standard concentration. Analyses will depend on the data, but will include univariate analyses across groups (eg, ANOVA) as well as multivariate approaches (eg, principal component analysis). Significance thresholds will be corrected for multiple hypothesis testing via Bonferroni. Data will be visualised using heatmaps, cluster plots and z-plots.
Macrophages
For macrophage analysis, one-way analysis of variance will be performed, followed by post hoc pairwise parametric testing.
Nutrigenomics
PBMC subsets (lymphocyte and monocytes) will be sorted and cDNA libraries prepared using QIAseq UPX 3′ transcriptome reagents before sequencing. Normalised gene expressions will be obtained through the CLC Genomics Workbench (Qiagen) and differentially expressed genes determined (false discovery rate-corrected p value<0.05) using the Limma package and performing partial least squares-discriminant analysis modelling to calculate their importance (variable importance in projection score). Functionally related genes will be identified and classified using pathway and analysis tools and the strongest indicators of response will be predicted by random forest area under the curve analysis.
Transcriptomic analysis will be conducted to compare RNA isolated from PBMCs from selected CD-TDI responders versus CD-TDI non-responders (n=8 per group). These will be selected as the best responders according to FCP and HBI at week 13. Sequencing libraries will be prepared using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (E7760) and the NEBNext Multiplex Oligos for Illumina (dual index primers set 1) (E7600) and sequencing performed on a NextSeq500 Sequencer (Illumina) at the Centre for Health Genomics and Informatics, University of Calgary (https://www.ucalgary.ca/dnalab/sequencing). For differential gene expression, parametric analysis with Sleuth will be conducted, using SLUG overexpression as the explanatory variable46 and a default filter function keeping any transcript with at least five reads in >47% of the samples. Transcripts passing the Wald test with Benjamini-Hochberg corrected p values (also known as false discovery rate) less than 0.05 will be considered to have been differentially expressed.
Differentially expressed genes will be analysed using pathway enrichment tools to obtain a global perspective of how the CD-TDI influences T cell and monocyte inflammatory gene expression.
Physical activity and sedentary time
Linear mixed model analysis will be used to investigate differences in the change in moderate/vigorous physical activity, steps and sitting time achieved between groups at the end of the intervention, after adjusting for valid accelerometer and inclinometer wear time and the number of valid wear days. To determine if physical activity and sedentary time are important covariates, stratified analyses (eg, active, inactive, sedentary) will be conducted when examining the effects of the intervention on clinical outcomes.
Biomarker identification and predictive modelling/patient stratification tool
A number of machine learning approaches will be considered and evaluated to identify biomarkers discriminating CD-TDI responders from CD-TDI non-responders and to develop a preliminary predictive model of clinical response to the CD-TDI. Parameters such as baseline cellular fatty acid profiles, PBMC gene expression and physical activity intensity, together with baseline taxonomic and functional microbiome profiles, will be analysed via machine learning-supported, supervised classification based on a single, clearly defined phenotype of response.
Baseline microbiome abundance profiles will be analysed, individually or in combination with clinical metadata, using methods suitable for compositional, high-dimensional and sparse data, to identify predictive features discriminating CD-TDI responders from CD-TDI non-responders. Due to the inherent data complexity, a variety of feature selection methods will be used, such as regularisation and filter-based methods, to select important readouts (microbiome, metabolome, cellular fatty acid, physical activity, gene expression) that will be employed to generate patient/disease stratification models and to develop models predictive of response to therapeutic diet.
Ethics and dissemination
Research ethics approval
The research protocol was approved by the Conjoint Health Research Ethics Board at the University of Calgary (approval number: REB19-0402) and the Health Research Ethics Board—Biomedical Panel at the University of Alberta (approval number: Pro00090772). Investigators will be made aware of any protocol amendments through regular study staff meetings, and any changes will be communicated to the funding body through twice-yearly reports.
Informed consent
The study coordinator will obtain informed consent from participants prior to their participation in the study.
Confidentiality and data management
Study staff will link participants to a unique alphanumerical identifier, saved in a master key on a password-protected server that requires multifactor authentication for access, and will enter all participant data into a secure data capture platform, through a password-protected computer. After study completion, only deidentified data will be used for analytic purposes. Only study investigators or their approved representatives will have access to the data. Once the research is completed, the data will be stored on a password-protected server for 5 years before it is destroyed according to organisational procedures.
The RD will meet virtually with the participants via telephone or a secure online platform accessed through a password-protected account to help protect the participants’ confidentiality. Meetings will not be recorded. The RD and study coordinator will know participants’ first and last names as they will be providing nutrition counselling or following up with participants.
Dissemination plan
The study results will be presented at national and international conferences and will be submitted for publication as abstracts and manuscripts. The study findings will also be shared on social media and educational material will be disseminated to stakeholder patient foundations.
Discussion
Experience with exclusion diets in patients with CD suggests that dietary interventions may be used to improve disease severity in subsets of patients, such as paediatric patients with mild-to-moderate luminal disease.15 However, the generalisability of these diets to adult patients, their sustainability and their effects across severity and disease phenotype are unknown. Furthermore, previous dietary RCTs have not used established mechanisms defined in patients with IBD to inform diet composition.
The planned RCT will determine whether a CD-TDI has an impact on clinical and biomarker evidence of inflammation and, moreover, will explore various mechanisms that could inform personalised diet therapy. The mechanistic outcomes that will be studied are broad and encompass clinical, dietary, genetic, microbiome, metabolomic and cellular details. To our knowledge, no other studies have explored the effects of diet on these types of readouts.
The multicentre approach to this study, along with broad outreach through social media and advertising on Crohn’s and Colitis Foundation website, will help us achieve adequate participant enrolment. Adherence to dietary interventions is a known challenge. To promote adherence to the CD-TDI, the study protocol includes frequent touchpoints with the RD to solve barriers to dietary adherence, as described in the Methods and analysis section. These touchpoints will be virtual to reduce the patient’s burden to attend visits. The recipes and food lists included in the study materials are designed to help patients more easily incorporate the CD-TDI into their lives.
This study will identify parameters associated with response to the CD-TDI to help us better understand and predict individual variations in response to the diet . Ultimately the research will provide clinical evidence for the potential of a viable, low-risk, adjunct treatment option for patients with mild-to-moderate CD for clinical and biochemical improvement and inform parameters which predict response to the CD-TDI.
Data availability statement
Data sharing not applicable as no datasets were generated and/or analysed for this study.
Ethics statements
Patient consent for publication
Acknowledgments
The authors would like to thank Munazza Yousuf, MD, Haley Pomreinke, RD, and Sandeep Kaur, BSc, for their contributions to the practical methods of the study, ethics and study organisation. The authors would also like to thank Rosica Valcheva, PhD, for her contributions to developing the process for the metabolomics analysis and Celeste Lavallee, RD, MSc, for her assistance in developing the manuscript.
References
Footnotes
Contributors MRaman, CM, LMT and SG conceptualised and designed the study. All authors developed or contributed to the study protocol design, critically reviewed and edited the manuscript, provided consultation on clinical data collection, and read and approved the final version of the manuscript. All authors will contribute to data analysis. MRaman developed the first draft of the manuscript. MRaman, CM, LAD and HJ assisted with recruitment.
Funding This work was supported by the Crohn’s & Colitis Foundation, Precision Nutrition in IBD program, grant number 672607.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.