Article Text

Abstract
Objective Tumour necrosis factor signalling via the receptor-interacting protein kinase 1 (RIPK1) pathway regulates colonic inflammation suggesting that RIPK1 inhibition may be a potential therapeutic target in ulcerative colitis (UC). This phase IIa, randomised, double-blind experimental medicine study investigated the safety, pharmacokinetics (PK), pharmacodynamics (PD) and preliminary efficacy of the RIPK1 inhibitor GSK2982772 in patients with active UC.
Design In part A, prior to a protocol amendment, one patient was randomised to receive GSK2982772 60 mg twice daily for 42 days. After the amendment, patients were randomised 2:1 to receive GSK2982772 60 mg or placebo three times daily for 42 days. In part B, all patients switched to open-label GSK2982772 60 mg three times daily for 42 days. Safety, PK, PD biomarkers, histological disease activity, clinical efficacy and quality of life were assessed at days 43 and 85.
Results Thirty-six patients were randomised (n=12, placebo/open-label GSK2982772; n=24, GSK2982772/open-label GSK2982772). Most adverse events were mild, with headache reported the most frequently across groups (placebo/open-label GSK2982772, n=2 (17%); GSK2982772/open-label GSK2982772, n=8 (33%)). GSK2982772 was well distributed into colonic tissue, with generally higher concentrations in colonic biopsy samples versus plasma. No apparent differences between treatment groups were observed for PD, histological disease activity, clinical disease activity or quality-of-life measures. At screening, all patients had Mayo endoscopic scores of 2 or 3. At day 43, no patients in the placebo/open-label GSK2982772 group achieved Mayo endoscopic scores of 0 or 1 vs 3/24 (13%) for GSK2982772/open-label GSK2982772. At day 85, 1/9 (11%) achieved scores of 0 or one for placebo/open-label GSK2982772 vs 3/22 (14%) for GSK2982772/open-label GSK2982772.
Conclusion GSK2982772 was generally well tolerated, with no treatment-related safety concerns identified. However, no significant differences in efficacy were observed between treatment groups, suggesting that GSK2982772 as monotherapy is not a promising treatment for patients with active UC.
Trial registration number NCT02903966.
- pharmacokinetics
- ulcerative colitis
- inflammatory bowel disease
- TNF
- colonic diseases
Data availability statement
Data are available on reasonable request. Within 6 months of this publication, anonymised individual patient data, the annotated case report form, protocol, reporting and analysis plan, data set specifications, raw dataset, analysis-ready dataset and clinical study report will be available for research proposals approved by an independent review committee. Proposals should be submitted to www.clinicalstudydatarequest.com. A data access agreement will be required.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Summary box
What is already known about this subject?
The receptor-interacting protein kinase 1 (RIPK1) pathway regulates colonic inflammation suggesting that RIPK1 inhibition may be a potential therapeutic target in ulcerative colitis (UC). GSK2982772 is a first-in-class oral, selective inhibitor of RIPK1.
What are the new findings?
GSK2982772 was well tolerated and, although well distributed into colonic tissue, there was no evidence of an effect of RIPK1 inhibition on disease activity.
How might it impact on clinical practice in the foreseeable future?
Inhibition of RIPK1 with GSK2982772 used as monotherapy is unlikely to affect disease activity to induce clinical improvement in UC.
Introduction
Ulcerative colitis (UC) is one form of inflammatory bowel disease characterised by chronic relapsing and remitting inflammation of the colon.1 The introduction of anti-tumour necrosis factor (anti-TNF) biologics, biologics that interfere with gut-specific lymphocyte trafficking, the interleukin 12 (IL-12)/IL-23 inhibitor ustekinumab and janus kinase inhibitors have improved the treatment landscape for patients with moderate to severe UC.2 3 However, there remains a high unmet need for more efficacious therapies. Oral therapies are often preferred and therapies with novel mechanisms of action are important for the treatment of patients with UC due to long-term efficacy limitations, the development of antidrug antibodies, and adverse effects associated with approved therapies.3 4
In response to engagement of the TNF receptor 1, receptor-interacting protein 1 (RIP1) exerts its signalling functions through both its catalytic kinase activity and as a scaffolding protein.5–7 Through its catalytic kinase activity, RIP1 plays an essential role in TNF-mediated necroptosis. By acting as a scaffolding protein, RIP1 facilitates other immune processes including TNF-mediated classical apoptosis and NF-κB (nuclear factor kappa B) signalling. Apoptosis and necroptosis have been implicated as drivers of inflammation in immune-mediated inflammatory diseases and histologically there is evidence for both in biopsies from patients with UC.8–10 Murine studies have suggested that colonic inflammation results from TNF signalling via the RIP kinase 1 (RIPK1) pathway.11 12 Additionally, bolus administration of TNF to mice results in necroptosis and severe intestinal inflammation that is RIPK1-dependent.13 Thus, RIPK1 is a potential therapeutic target in UC.
GSK2982772 (5-Benzyl-N-[(3S)−5-methyl-4-oxo-2,3-dihydro-1,5-benzoxazepin-3-yl]−1H-1,2,4-triazole-3-carboxamide) is a first-in-class oral, selective inhibitor of RIPK1 that binds to an allosteric pocket of the RIPK1 kinase domain to inhibit RIPK1-mediated cell death and cytokine production,14 and its safety and tolerability have been demonstrated at doses up to 120 mg twice daily in healthy volunteers.15 The objective of this two-part experimental medicine study was to examine the safety, pharmacokinetic (PK), pharmacodynamic (PD) and preliminary efficacy of GSK2982772 (up to 60 mg three times daily) at 43 and 85 days in patients with active UC.
Participants and methods
Study design
This was a phase IIa, multicentre, randomised double-blind (sponsor-unblinded), placebo-controlled study with an open-label extension to investigate the safety and tolerability, PK, PD and preliminary efficacy of GSK2982772 in patients with active UC (NCT02903966). The study was conducted at 23 centres in 7 countries between 15 November 2016 and 17 June 2019, and consisted of a 30-day screening period, a two-part treatment period (each part 42 days) and a 28-day follow-up period.
An interactive web response system was used for the randomisation of patients and the randomisation list was generated in RandALL. All study staff and patients were blinded to treatment assignment. In part A, one patient was randomised to receive GSK2982772 60 mg twice daily. Patients were initially assigned to either GSK2982772 60 mg orally twice daily or placebo twice daily, which was an inactive, white, round, film-coated tablet matched in appearance to active tablet. Following a protocol amendment effective 20 April 2017, the dosing regimen was changed and patients in part A were randomised 2:1 to receive GSK2982772 60 mg (n=24) or placebo (n=12) three times daily rather than twice daily. In part B (after the 42-day treatment period for part A), all patients switched to open-label GSK2982772 60 mg three times daily for an additional 42 days.
Patients
To be eligible for the study, patients needed to be between 18 and 75 years of age with UC and a total Mayo score of ≥3 points and endoscopy subscore of 2–3 at screening16 17 despite stable dosing of concurrent treatment under the following conditions: oral 5-aminosalicylates (≥2.4 g/d mesalamine) for at least 4 weeks prior to first dose of study drug, purine analogues or methotrexate for at least 12 weeks prior to first dose of study drug, or low-dose corticosteroids (≤20 mg prednisolone or equivalent) for 2 weeks prior to sigmoidoscopy. Dosing of concurrent treatment was required to remain stable until the end of the study. Eligible patients were to be either naïve to any biological therapies used for UC or, alternatively, to have discontinued anti-TNF biological therapy for reasons other than primary efficacy nonresponse. Exposure to a single biological agent in the context of a clinical trial was also acceptable.
Key exclusion criteria included a diagnosis of indeterminate colitis, Crohn’s disease, infectious colitis or ischaemic colitis, fulminant UC or UC limited to the rectum (disease extent <15 cm from the anal verge), previous small bowel or colonic surgery (with exception of appendectomy), and histological evidence of colonic dysplasia or bowel stricture. Patients with fistulae or known symptomatic stenosis of the intestine, toxic megacolon, an active infection or history of infections (including a positive Clostridium difficile toxin test), or active/previous colonic cytomegalovirus infection were also excluded. Patients with a history of suicidal ideation behaviour as measured using the Columbia Suicide Severity Rating Scale (C-SSRS) or history of attempted suicide were not eligible.18
Study end points and assessments
Safety and tolerability were the primary end points of the study. Secondary endpoints included efficacy measures, histological disease activity, PK, and PD effect on biomarkers of disease activity. Change from baseline in Inflammatory Bowel Disease Questionnaire scores was an exploratory endpoint.
Clinical laboratory values, vital signs and 12-lead ECG monitoring were assessed at screening, day 1 and every 2 weeks thereafter until day 85 (predose on days 1 and 43). Patients also completed the C-SSRS predose on days 1, 43 and 85. The investigator or site staff and an internal GSK Safety Review Committee (which included members of the GSK2982772 project team) monitored adverse events (AEs) and reviewed blinded safety data. Clinical laboratory parameters were assessed every 2 weeks and AEs were assessed weekly throughout the study.
The Inflammatory Bowel Disease Questionnaire (IBDQ), a 32-item health-related quality-of-life instrument specific to inflammatory bowel disease,19 was used to evaluate general activities of daily living, intestinal function, social performance, personal interactions and emotional status. Questionnaires were completed by patients before any other clinical assessments on days 1 and 43 (predose) and day 85.
Efficacy assessments included the total Mayo score, a 12-point scoring system used to assess UC disease activity based on stool frequency, rectal bleeding, endoscopic appearance, and the Physician’s Global Assessment of Disease (PGA)20; and the Ulcerative Colitis Endoscopic Index of Severity (UCEIS), which assesses disease activity based on endoscopic vascular pattern, bleeding, erosions and ulcerations.21 Total Mayo score, partial Mayo score (stool frequency, rectal bleeding and PGA), 3-domain Mayo score (stool frequency, rectal bleeding and mucosal appearance), Mayo endoscopic subscore and the UCEIS were used to assess disease activity at screening, day 43 (predose) and day 85.
A total of 6–12 random endoscopic biopsies were obtained throughout the inflamed sigmoid during sigmoidoscopy at screening, day 43 (predose) and day 85, and were divided for histological, PK, target and pathway engagement, and gene expression assessments. For histological assessment, samples were stored in 20 mL formalin solution directly after biopsy. Formalin-fixed paraffin-embedded blocks were prepared and samples were batch shipped to Robarts (Mount Sinai Services; Toronto, Canada) for processing and analysis. Samples were scored using Geboes index,22 the modified Riley score,23 and Robarts histology index,24 which are histological scoring instruments for the evaluation of disease activity in UC.
Predose plasma concentrations of GSK2982772 were measured on days 43 and 85 (for comparison with predose colon tissue concentrations). In addition, postdose (1, 2, 4 and 6 hours) plasma concentrations were assessed on days 1 and 43. Plasma and biopsy samples were analysed using a validated analytical method based on protein precipitation with [2H313C] GSK2982772 as an internal standard, followed by high-performance liquid chromatography/mass spectrometry analysis.
PD biomarkers included C reactive protein (CRP) (predose on days 1 and 43) as a part of chemistry laboratory samples and faecal calprotectin (FCP). On day 1 and every 2 weeks thereafter, stool samples were collected into a 30 mL plastic stool container and stored at −20°C before being shipped the same day on dry ice to Q2 Solutions.
Statistical analysis
This exploratory experimental medicine study was not powered to test predefined differences in clinical efficacy or biomarkers. The safety population consisted of all patients who received at least 1 dose of study treatment (safety, efficacy and biomarker analyses).
Safety, efficacy, PK and biomarker data were summarised for part A according to the treatment received and, as appropriate, were summarised for parts A and B of the study combined. Comparisons between treatment groups were conducted for clinical efficacy, histological and biomarker assessments if deemed appropriate using a mixed model repeated measures model, adjusting for the following covariates: treatment, visit, log-transformed baseline value and the treatment by visit and log-transformed baseline value by visit interactions.
Generalised estimating equations modelling was planned to compare the proportion of patients achieving Mayo endoscopy remission (scores of 0 or 1), Mayo clinical remission or 3-domain Mayo clinical remission between treatment groups at each time point. However, statistical analyses were not performed due to convergence issues.
IBDQ item responses were summed up for a total score and averaged among four dimensions: bowel function (10 items), systemic symptoms (5 items), social function (5 items) and emotional status (12 items).
Descriptive statistics were used to summarise GSK2982772 predose colon tissue concentrations by day and GSK2982772 plasma concentrations by day and nominal sampling time.
Results
Disposition and demographics
A total of 36 patients were enrolled—12 in the group treated with placebo for the first 42 days followed by open-label GSK2982772 for 42 days (placebo/OL’772 group), and 24 in the group treated with GSK2982772 for the first 42 days followed by open-label GSK2982772 for 42 days (GSK2982772/OL’772 group). All 36 patients received treatment and were included in the safety population (online supplemental figure 1). All 24 of the patients in the GSK2982772/OL’772 group and 10 patients in the placebo/OL’772 group were included in the PK population. One patient withdrew from the placebo/OL’772 group due to an AE (worsening of UC) and 2 patients withdrew from the GSK2982772/OL’772 group (one withdrew consent, one due to lack of efficacy). Baseline demographics and UC characteristics were generally balanced between groups (table 1). The mean age of patients in the placebo/OL’772 group was older than the GSK2982772/OL’772 group and the ratio of men to women was 2:1 in the GSK2982772/OL’772 group vs 1:1 in the placebo/OL’772 group. Mean (SD) FCP level at baseline was 1101 (1517) UG/G in the placebo/OL’772 group compared with 1678 (2327) UG/G in the GSK2982772/OL’772 group. No patients in the placebo/OL’772 group and 7 in the GSK2982772/OL’772 group were using immunosuppressants at baseline.
Supplemental material
Patient demographics and baseline characteristics (safety population)
Safety and tolerability
AEs were reported in 75% of patients in both treatment groups (table 2). The most frequently reported AE in both treatment groups was headache (placebo/OL’772, n=2 (17%); GSK2982772/OL’772, n=8 (33%)). In the placebo/OL’772 group, 58% of AEs were reported during part A (while receiving placebo), including 1 AE of headache. Other AEs reported in >10% of participants in the GSK2982772/OL’772 group included nasopharyngitis (5 (21%)) and nausea, abdominal pain and upper abdominal pain in 3 (13%) patients each. Drug-related AEs were reported in 2 (17%) patients in the placebo/OL’772 group (both during part A) and 5 (21%) in the GSK2982772/OL’772 group.
Adverse events reported in >10% in any treatment group (safety population)
One patient in the placebo/OL’772 had a serious AE of worsening UC of moderate intensity on day 11 (while taking placebo), which led to withdrawal from the study. In the GSK2982772/OL’772 group, 2 patients had serious AEs (one with bladder cancer and one with a comminuted humerus fracture after a fall). No serious AEs were related to study treatment as judged by the investigator.
Apart from one patient in the GSK2982772/OL’772 group who experienced a QTcF increase between 450 and 480 ms, there were no clinically significant ECG changes reported. No clinical chemistry or haematology values of potential clinical importance were reported and no liver monitoring or stopping events occurred. No suicidality was reported during either part of the study.
Pharmacokinetics
Median GSK2982772 concentrations from sigmoid biopsies obtained predose on day 43 (88.0 ng/mL) were of similar magnitude to the corresponding plasma concentrations (58.4 ng/mL; online supplemental table 1).
PD and biomarker results
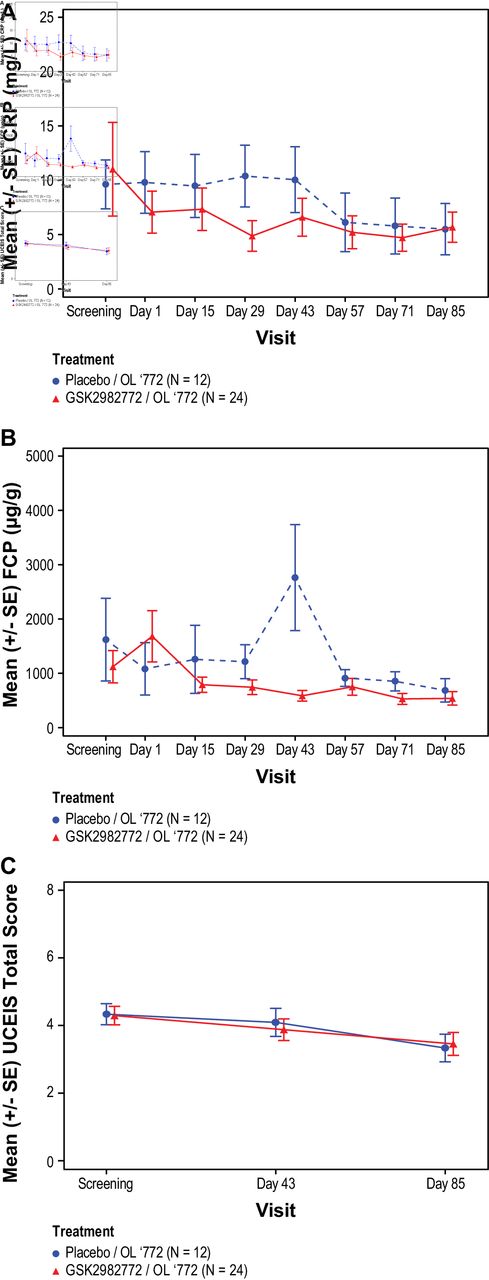
On day 1, predose mean (SD) CRP levels were 9.8 (9.82) in the placebo/OL’772 group and 7.1 (9.47) in the GSK2982772/OL’772 group. A reduction in CRP levels was observed in the GSK2982772/OL’772 group as compared with the placebo/OL’772 group on days 15, 29, and 43 (day 43 LS mean (SE): placebo/OL’772, 1.06 (1.854); GSK2982772/OL’772,–0.64 (1.251)) (online supplemental table 2). However, the CRP values were similar in both treatment groups on days 57, 71 and 85 (figure 1A).

Mean Ulcerative Colitis Endoscopic Index of Severity (UCEIS) over time by treatment group. (A) Mean CRP levels; (B) mean faecal calprotectin levels; (C) mean UCEIS total score. Placebo/OL’772 patients received placebo until day 43 when all patients switched to open-label GSK2982772 60 mg three times daily. GSK2982772/OL’772 patients received GSK2982772 60 mg three times daily until day 43 when all patients switched to open-label GSK2982772 at the same dose. CRP, C reactive protein; FCP, faecal calprotectin.
The FCP values were similar between treatment groups (online supplemental table 2) except for day 43 (Geometric LS mean (%CVb): placebo/OL’772, 1.90 (40.7); GSK2982772/OL’772, 0.44 (27.1)) when a steep increase associated with high variability of the FCP data was noted in the placebo/OL’772 group (online supplemental table 3, figure 1B).
Histological disease activity
No reduction in histological disease activity was observed in either treatment group on day 43. The adjusted mean change from baseline in modified Riley scores was 0.4 (0.842) in the placebo/OL’772 group and 0.4 (0.558) in the GSK2982772/OL’772 group. Small, non-clinically relevant reductions in Robarts histology index scores from baseline to day 85 were noted in both treatment groups (placebo/OL’772,–2.51 (3.377); GSK2982772/OL’772,–2.96 (2.171)). Mean Geboes index scores were unchanged in both treatment groups on days 43 (placebo/OL’772, 1.04 (1.847); GSK2982772/OL’772, 0.28 (1.223)) and 85 (placebo/OL’772, –0.67 (1.981); GSK2982772/OL’772, –1.47 (1.286)) (figure 2).

{kind=link}
{kind=link}
Mean modified Riley score, Geboes index and Robarts histology index over time by treatment group. (A) Modified Riley score; (B) Geboes index; (C) Robarts Histology Index (RHI). Placebo/OL’772 patients received placebo until day 43 when all patients switched to open-label GSK2982772 60 mg three times daily. GSK2982772/OL’772 patients received GSK2982772 60 mg three times daily until day 43 when all patients switched to open-label GSK2982772 at the same dose.
Clinical efficacy
The LS mean (SE) change from baseline at day 43 for total Mayo score was −1.42 (0.656) for the placebo/OL’772 group and −1.75 (0.443) for GSK2982772/OL’772. At day 85, observed decreases were similar in both treatment groups (placebo/OL’772, –3.20 (0.895); GSK2982772/OL’772,–3.16 (0.583)). Changes in partial Mayo score, and 3-domain Mayo score were also similar at days 43 (placebo/OL’772, –0.99 (0.527); GSK2982772/OL’772, –1.36 (0.356)) and 85 (placebo/OL’772, –2.32 (0.678); GSK2982772/OL’772,–2.19 (0.440)) regardless of randomised treatment group (online supplemental table 2, online supplemental figure 2). The percentage of patients achieving Mayo clinical response (defined as ≥3 points or ≥30% improvement from baseline in the total Mayo score, along with a decrease in the rectal bleeding subscore of ≥1 point) on day 85 for the placebo/OL’772 was 5/9 (56%) and 11/22 (50%) for GSK2982772/OL’772. One of 9 (11%) patients in the placebo/OL’772 and 2/22 (9%) in the GSK2982772/OL’772 achieved Mayo remission (defined as total Mayo score of ≤2 points, with no individual subscore exceeding 1 point). Three-domain Mayo clinical response or remission scores also increased over time and were similar between treatment groups with 6/9 (67%) in the placebo/OL’772 group and 12/22 (55%) in the GSK2982772/OL’772 at day 85 achieving a clinical response (table 3).
Patients achieving Mayo response and remission and UCEIS remission (safety population)
Reduction in UCEIS total scores was similar in both the treatment groups at days 43 (placebo/OL’772, –0.24 (0.428); GSK2982772/OL’772, –0.42 (0.289)) and 85 (placebo/OL’772, –0.84 (0.495); GSK2982772/OL’772, –0.82 (0.318); online supplemental table 2 and figure 1C). The percentage of patients achieving UCEIS response and remission increased over time and was similar between treatment groups (day 85: placebo/OL’772, 1/9 (11%); GSK2982772/OL’772 3/22 (14%)) (table 3). The mucosal appearance as assessed by the Mayo endoscopic subscore was similar between treatment groups. At day 43, no patients in the placebo/OL’772 group and 3/24 (13%) in the GSK2982772/OL’772 group achieved Mayo endoscopic scores of 0 or 1 indicating remission. The percentage of patients that achieved endoscopic scores of 0 or 1 at day 85 was 1/9 (11%) in the placebo/OL’772 group and 3/22 (14%) in the GSK2982772/OL’772 group (online supplemental table 4).
Quality-of-life measures
Patients in the placebo/OL’772 group had lower IBDQ scores at baseline relative to the GSK2982772/OL’772 group. The increase in IBDQ scores at day 43 (placebo/OL’772, 28.40 (8.274); GSK2982772/OL’772, 20.62 (5.668)) and at day 85 (placebo/OL’772, 41.72 (9.439); GSK2982772/OL’772, 37.72 (6.566)) was similar between treatment groups (online supplemental figure 3).
Discussion
Treatment with GSK2982772 60 mg three times daily for 43 days or 85 days was generally well tolerated. GSK2982772 was well distributed into colonic tissue, as shown by the similar GSK2982772 concentrations in the colonic biopsy samples and plasma. In spite of this, there was no evidence of an effect of RIPK1 inhibition on disease activity. For instance, there were no apparent differences in the change of CRP levels comparing patients treated with GSK2982772/OL’772 to placebo/OL’772. It should be noted that the placebo/OL’772 group, had a relatively smaller sample size compared with the GSK2982772/OL’772 group and that FCP values at baseline were highly variable. The steep increase observed in the placebo/OL’772 group at day 43 might be explained by one subject who had a large increase from baseline at day 43, likely reflecting a flare which is characteristic of the natural course of disease for patients with ulcerative colitis25 and impacting the group outcome.
Consistent with the results for CRP and FCP levels, small improvements of similar magnitude in histological disease activity were observed in both treatment groups. Changes in clinical efficacy measures and endoscopy scores to assess UC disease activity in response to treatment with GSK2982772 60 mg three times daily were also similar between treatment groups. Additionally, IBDQ scores increased over time, suggesting patients’ quality of life improved similarly in both treatment groups. A recent meta-analysis suggested that there is a meaningful placebo effect on HRQoL as reflected in IBDQ scores, which should be considered in clinical trials.26 The HRQoL improvements in the current study may be explained by this placebo effect or simply by the relapsing and remitting nature of ulcerative colitis over the course of the study.
Short treatment duration, treatment compliance with a three times daily regimen, dose level, insufficient concentrations of GSK2982772 to inhibit RIPK1 activity or biological redundancy could explain this lack of response. Taken together, these results do not support the notion that RIPK1 inhibition alone at the level achieved in this study could translate into transformative improvement of UC.27–29
Other limitations to the study include the relatively small sample size and that baseline information regarding disease severity, including Montreal classification, was not collected. The impact of disease heterogeneity between treatment groups may have affected outcomes. It is possible that statistically significant improvements would have been apparent with a larger sample size. However, if treatment effects were transformative, they would also be expected to be apparent in a smaller experimental medicine study, even given the above limitations.
Patients’ prior treatment history was not captured at baseline and, as treatment with biological therapy may indicate a population that is more refractory to treatment effects, results may have been affected. Finally, no patients in the placebo/OL’772 group were taking immunosuppressants at baseline vs 7/24 (29%) in the GSK2982772/OL’772 group, which could indicate a more difficult group to treat. Despite these limitations and preclinical evidence that RIPK1 may be a promising therapeutic target, current findings suggest that a transformative effect is unlikely with GSK2982772 used as monotherapy to inhibit RIPK1 activity and to induce clinical improvement in UC.
Conclusions
In conclusion, this study did not identify any significant safety issues and the treatment of patients with active UC using the RIPK1 inhibitor GSK2982772 60 mg three times daily for 43 or 85 days was generally well tolerated. However, at the exposure levels evaluated, the agent does not lead to differences in measures of histological disease activity or clinical efficacy compared with placebo. RIPK1 may not be a promising therapeutic target in UC when GSK2982772 is used as monotherapy.
Supplemental material
Data availability statement
Data are available on reasonable request. Within 6 months of this publication, anonymised individual patient data, the annotated case report form, protocol, reporting and analysis plan, data set specifications, raw dataset, analysis-ready dataset and clinical study report will be available for research proposals approved by an independent review committee. Proposals should be submitted to www.clinicalstudydatarequest.com. A data access agreement will be required.
Ethics statements
Ethics approval
The study was approved by the medical ethics committee at every participating institution and was conducted according to the recommendations of Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent to participate in the study.
Ethics Committee/Institutional Review Board List (Centre Numbers) Germany: Ethik-Kommission der Landesaerztekammer Hessen, Im Vogelsgesang 3, Frankfurt, Hessen, 60488, Germany (228191, 228192, 228193). Netherlands: Medisch Ethische Commissie, Meibergdreef 9, Amsterdam, 1105 AZ, Netherlands (224520). Poland: Komisja Bioetyczna przy Okregowej Izbie Lekarskiej w Lodzi, ul. Czerwona 3, Lodz, 93–005, Poland (227890, 235633, 227795). Russian Federation: Federal medical biophysical centre n.a. A.I. Burnazyan of FMBA of Russia, 23, Marshala Novikova street, Moscow, 123098, Russian Federation (237613); EC of Ufa City Clinical Hospital, #21, 3 Lesnoy pr, Ufa, 450071, Russian Federation (235576); EC of Stavropol State Medical University 310 Mira street, Stavropol, 355017, Russian Federation (235413); Ethics Comitee of Saint Petersburg State Budget Institution of Healthcare 'City Hospital #26', Ulitsa Kostyushko, 2, Saint Petersburg, 196247, Russian Federation (235283); Rostov Sate Medical University of RosZdrav, State Educational Institution of High Professional, 29, Nakhichevansky pereulok, Rostovon-Don, 344022, Russian Federation (235135); LEC of LLC 'Astarta', Galernyy passage, 5, Saint-Petersburg, 199226, Russian Federation (238410). Sweden: Etikprövningsmyndigheten, Box 2110, 750 02 Uppsala, Sweden (222728, 222727, 222729). UK: West Midlands-Edgbaston Research Ethics Committee The Old Chapel, Royal Standard Place, Nottingham, NG1 6FS, UK (224368, 224687); NRES Committee West Midlands-Coventry & Warwickshire The Old Chapel, Royal Standard Place, Nottingham, NG1 6FS, UK (235414, 224304). USA: Copernicus Group-Institutional Review Board Suite 200, 5000 Centre Green Way, Cary, North Carolina, 27513, USA (231423, 237076); Western Institutional Review Board-Copernicus Group Institutional Review Board, Suite 100, One Triangle Drive, Research Triangle Park, North Carolina, 27709, United States (231295, 223623, 225027); NYU School of Medicine Institutional Review Board, 1 Park Avenue, sixth Floor, New York, New York, 10016, USA (223627); Western Institutional Review Board, 1019 39th Avenue SE, Puyallup, Washington, 98374, USA (223624); Copernicus Group, Suite 200, 5000 Centre Green Way, Cary, North Carolina, 27513, USA (238304).
Acknowledgments
All listed authors meet the criteria for authorship set forth by the International Committee for Medical Journal Editors. Editorial support (Allyson Lehrman, DPM and Sarah Hummasti, PhD) with assembling tables and figures, collating author comments, copyediting, fact checking and referencing and graphic services were provided by AOIC, LLC and were funded by GlaxoSmithKline. The authors thank the patients and study investigators for their participation in the study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors KW, SW, MP, CW, DJT, SWB and PPT contributed to the conception or design of the study; SW, KB, MP, MB, CW, DJT, SH and KA-B contributed to the acquisition of the data; KW, NS, MP, MB, DJT, SH, KA-B and PPT contributed to the data analysis or interpretation. All authors provided critical review and final approval of the publication and agree to be accountable for the work.
Funding This study (ClinicalTrials.gov Identifier: NCT02903966) was funded by GlaxoSmithKline (GSK) and all authors were employees at the time of the study. GSK authors were involved in the study design, data collection and analysis, and decision to publish.
Competing interests NS, MP, MB, CW, DJT, SWB and KA-B are employees of and hold equity in GlaxoSmithKline (GSK). KW, SB, SW, KB, SH and PPT are former employees of and may hold GSK stocks.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.