Article Text

Abstract
Introduction Fatigue is the most commonly reported symptom of the liver disease primary biliary cholangitis (PBC). It affects 40%–80% of patients, has no effective treatment and is associated with heightened mortality risk. The pathogenesis is unknown, but muscle bioenergetic abnormalities have been proposed to contribute. Directly observed exercise has been shown to attenuate symptoms in small groups; however, due to the rare nature of the disease, home-based interventions need to be evaluated for feasibility, safety and efficacy.
Methods and analysis This is a phase 1/pilot, single-arm, open-label clinical trial evaluating a novel home-based exercise programme in patients with PBC with severe fatigue. Forty patients with moderate-severe fatigue (PBC40 fatigue domain score >33; other causes of fatigue excluded) will be selected using a convenience sampling method. A 12-week home-based exercise programme, consisting of individualised resistance, aerobic exercises and telephone health calls (first 6 weeks only), will be delivered. Measures of fatigue (PBC40 fatigue domain; fatigue impact scale), quality of life, sleep (Epworth Sleep Score), physical activity, anxiety and depression, aerobic exercise capacity (incremental shuttle walk test; Duke Activity Status Index) and functional capacity (short physical performance battery) will be assessed at baseline and at 6 and 12 weeks following the intervention.
Ethics and dissemination The protocol is approved by the National Research Ethics Service Committee London (IRAS 253115). Recruitment commenced in April 2019 and ended in March 2020. Participant follow-up is due to finish by December 2020. Findings will be disseminated through peer-reviewed publication, conference presentation and social media.
Trial registration number NCT04265235.
- primary biliary cirrhosis
- quality of life
- autoimmune liver disease
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Primary biliary cholangitis (PBC) is a chronic, immune-mediated liver disease characterised by progressive reduction in bile flow (cholestasis) that leads to biliary fibrosis and eventual cirrhosis.1 The exact disease mechanisms are unknown but considered multifactorial, encompassing inherited traits and ill-defined environmental factors.2 Incident rates suggest c35 per 100 000 individuals are affected with PBC, of whom 90% are women aged between 30 and 65 years at the time of diagnosis.3
Debilitating, refractory fatigue is one of the most commonly reported symptoms, affecting between 40% and 80% of patients.4 Although not an indicator of liver disease severity, fatigue is pronounced among individuals of young presenting age and is associated with worse liver transplant-free survival, reduced physical activity and poorer quality of life (QoL) for patients. While a consensus biological explanation is lacking, preclinical data have identified central and peripheral components, alongside suppressed anaerobic threshold during physical exertion.5 6 Notably, patients with PBC suffering from fatigue report that it is not a single episode of activity they find difficult, rather the incapacity to perform exercise over a sustained period of time. This relates to excessive acidosis building up in peripheral muscle coupled with delayed pH recovery, which tends to manifest after only a minimal duration of physical exertion, compared with non-fatigued patients with PBC and healthy control subjects.5 7 Importantly, this pathology can be reversed through repeated single exercise episodes, suggesting that the capacity to improve muscle bioenergetics is retained.5 Most strikingly, supervised graded exercise intervention demonstrates significant and quantifiable improvements in fatigue severity, together with symptom reduction in domains related to emotional and social dysfunction.5
While patients with liver disease report positive attitudes towards the benefits of supervised exercise, they admit to lack of confidence were they to initiate such activity themselves.5 8 Moreover, those living with fatigue have a natural inclination towards decreased levels of physical activity in response to symptoms.9 Similar to other chronic diseases, individuals are more likely to disengage, become socially isolated and increase sedentary living due to fear of exacerbating fatigue if correct guidance is not provided.10 The resulting physical inactivity is a catalyst for muscular degeneration, with reduced exercise tolerance and functional capacity, thereby increasing fatigability.9 10 This ‘fatigability’ further exacerbates fear and sedentary living, leading to a vicious cycle of debilitating decline.9 The consequence of this is further physical inactivity and muscular degeneration, a reduction in oxygen-handling (oxygen consumption peak) and functional capacity, and subsequently the speed at which the muscle fatigues.9–11
Access to healthcare professionals appears to reduce self-reported fatigue and positively influence the attitudes towards exercise and the long-term benefits on overall QoL.12–14 Moreover, home-based exercise programmes (HBEPs) performed at moderate intensity (rate of perceived exertion (RPE) 12–14) have been shown to be safe and improve functional capacity in patients with end-stage liver disease.15 However, there are no studies that explore the feasibility and efficacy of a moderate-intensity HBEP on improving fatigue or functional capacity in patients with PBC.
Prior to performing a dedicated randomised controlled trial (RCT), a pilot feasibility study is required to determine whether intervention can be conducted within patients’ homes, determine whether a larger programme is possible and highlight preferential design features. To this effect, our aim is to conduct a single-centre trial of a structured HBEP, which attenuates fatigue associated with PBC. Efficacy assessment will be through validated health-related quality of life (HRQoL) measures, and fatigue-specific and functionality assessment tools.
Methods and analysis
Study design overview
This is a single-arm, single-centre feasibility trial of a graded HBEP for the treatment of chronic fatigue in patients with PBC. Patients will be recruited through the Liver Unit based at the University Hospitals Birmingham (UHB) National Health Service (NHS) Foundation Trust, UK. An individualised, graded HBEP will be delivered for 12 weeks to investigate safety, adherence and treatment efficacy (figure 1). Fatigue, HRQoL, functional capacity and sleep quality will be recorded at baseline and at 6 and 12 weeks following initial assessment.

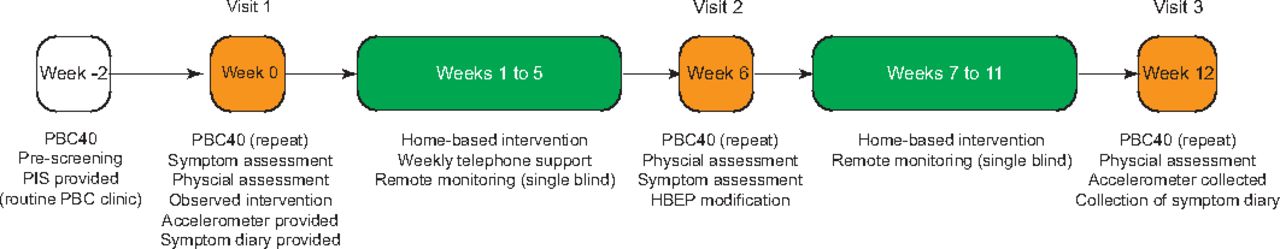{kind=link}
Study overview. Patients with moderate-severe fatigue (PBC40 fatigue domain score >33) will be identified from clinic and PBC40 fatigue assessment completed. Eligible participants will be invited to attend a dedicated screening visit within 2 weeks and a repeat PBC40 questionnaire completed. After obtaining consent, the investigator will perform full physical and symptom assessment of the trial participant and demonstrate the intended intervention. The trial participant will then be observed while performing the aforementioned intervention and will be provided a symptom diary and single-blinded GeneActiv accelerometer for home use (daily activity monitor readings will be captured remotely by the investigator, but not visible by the participant). Thereafter, the participant will be instructed to perform a tailored, daily HBEP by the investigators (liver physiotherapist and personal trainer). Weekly telephone support will be provided in the first 6 weeks (interval between visit 1 and visit 2), together with modifications to the exercise programme as needed. At week 6, the trial participant will be invited for an interim assessment, followed by another 6 weeks of intervention. Weekly telephone support will be withdrawn between weeks 6 and 12 (end of the study). Assessment of the primary efficacy measure will be performed at week 12 (end of study visit). HBEP, home-based exercise programme; PBC, primary biliary cholangitis; PIS, patient information sheet.
Sample and selection
Patients attending the dedicated PBC clinic will be presented with a PBC40 questionnaire, which is a robustly validated HRQoL measure specifically for application in PBC.16 Given that some patients experience fatigue by virtue of having advanced disease, our study will exclusively target individuals who have clinically significant symptoms although with fully compensated liver function. Patients with clinically significant fatigue (defined by a PBC40 fatigue domain score >33) will be asked to complete a second questionnaire remotely, 14 days after initial assessment.16 17 Patients with persistently elevated scores (in the absence of other contributory causes) will be deemed eligible for trial participation. Additional criteria for trial entry include the following.
Inclusion criteria
≥ 18 years old.
A confirmed diagnosis of PBC, in keeping with international guidelines.18
Exclusion criteria
Decompensated liver disease, as evident by the presence of ascites, hepatic encephalopathy (any grade) and history of variceal bleeding.
Total bilirubin of >50 μmol/L, in the absence of Gilbert’s syndrome.
Prior liver transplantation.
On the waitlist for or likely to need referral for liver transplantation in the next 6 months (judged by the principal investigator (PI)).
Refractory pruritus (judged by the PI).
No dual-energy X-ray absorptiometry scan within 5 years.
Untreated osteoporosis.
Cardiovascular instability (judged by PI).
Untreated hypovitaminosis or anaemia.
Untreated hypothyroidism.
Untreated coeliac disease.
WHO performance status ≥3.
History of unexplained falls.
Neither patient nor next of kin are English speaking.
Refusal or lack of capacity to give informed consent.
Participation in another interventional trial for PBC in the last 3 months.
Concomitant excess alcohol consumption (>14 units for women; >21 for men).
Concomitant liver disease of another aetiology, including (but not limited to) viral hepatitis, autoimmune hepatitis, primary sclerosing cholangitis, alcohol-induced liver disease, non-alcoholic fatty liver disease, Wilson disease, alpha-1-antitrypsin deficiency, Budd-Chiari syndrome, hereditary haemochromatosis, drug-induced liver injury and IgG4-related disease.
Intercurrent active or latent infection.
Intercurrent immunocompromised state.
Concurrent chemotherapy.
Active malignancy (other than non-melanomatous skin cancer).
Once screened and deemed eligible, patients will be contacted via telephone to discuss their involvement within the study, and a patient information sheet will sent sent by the chief investigator (CI) or co-investigator (Co-I). If individuals agree to take part, an appointment will be arranged for written consent, baseline assessment and an initial supervised exercise session.
Study visits
Patients who suffer from PBC attend regular clinic appointments as part of routine clinical care. Where possible, all study visits will be conducted on the same day as routine clinic appointments to reduce the burden of hospital attendance and travel.
The study requires three separate attendances to the clinic: at week 0 (visit 1—baseline), week 6 (visit 2) and week 12 (visit 3—end of study) (figure 1). At each visit, the assessment of fatigue, HRQoL, functional capacity and sleep quality will be quantified.
Visit 1
At week 0 (baseline), fatigue will be quantified using the PBC40 fatigue domain and Fatigue Impact Scale (FIS).19 Additional metrics recorded at baseline will include the following:
Remaining components of the PBC40 assessment tool.16
The Chronic Liver Disease Questionnaire (CLDQ).20
The Hospital Anxiety and Depression Scale (HADS)21
The Epworth Sleep Score (ESS).22
Measures of functional and aerobic capacity:
Baseline assessment will also capture anthropometric data (age, sex, weight, height and body mass index), details of PBC-specific treatments (including ursodeoxycholic acid (UDCA), obeticholic acid, fibric acid derivatives, bile acid sequestrants, rifampicin, selective serotonin reuptake antagonists, opioid antagonists), dietetic input (eg, use of protein supplements) and laboratory data (liver biochemistry, renal function, thyroid function, vitamin D levels, haemoglobin A1c, full blood count and clotting profile).
Intervention and monitoring
Participants will be provided with a moderate-intensity, low-impact individualised HBEP dependent on index measures of physical activity, performance status and confidence with intervention during visit 1. Participants will be advised to exercise within a Borg RPE of 12–14, as this has been shown to correlate with anaerobic thresholds in healthy individuals and improve functional capacity in patients with end-stage liver disease.15 27 In addition, each participant will be given a GENEActiv wrist accelerometer in a single-blind manner (which will provide retrospective feedback on activity to the investigators but not the participants) to wear 24 hours a day during the 12-week study period. The GENEActiv will encompass assessment of physical activity and sleep quality. In parallel, a paper-based patient diary will be provided to record HBEP targets, alongside a copy of their specific programme.15
Home-based exercise program
Following discussions with our patient and public involvement group, it was recommended that all interventions should include functional movements, require no specialist equipment, that exercise instructions should be supported with written information and the overall commitment should not involve more than three sessions per week. Therefore, at initial assessment, participants will be taught functional resistance exercises that they can complete at home (table 1 and online supplemental appendix 1)—as previously described.15 Entry levels will be determined depending on individual physical capacity and confidence. Each participant will be observed completing their specific exercise programme during visit 1, under the supervision of an accredited specialist physiotherapist and a qualified personal trainer. Patient technique, safety and confidence in completing each exercise will be assessed, as well as ensuring they can maintain an RPE of 12–14 throughout the exercise session. Participants will be advised to complete functional resistance exercises 2–3 times per week (depending on capability) as this has been shown to correlate with physiological change and will be provided with written information for each exercise to guide completion at home. Although patients will not be directly supervised while conducting exercises at home, remote monitoring through assessment of accelerometer data and weekly telephone calls will be conducted during this time.15
Supplemental material
Exercise levels prescribed for participants
The RPE is a psychophysical tool that is widely used to assess the perception of effort during exercise across a multitude of health conditions and positively correlates with both heart rate and blood lactate.27 Given its simplicity, acceptance within healthcare and assurance that it correlates with physiological change, the use of RPE during execution of the field test and HBEP was deemed an appropriate choice. This will also allow for an individualised approach and inclusivity of participants despite baseline functional ability. Supplementary telephone calls will allow for remote progression and regression of exercises and monitoring of adverse symptoms including exertion-related chest discomfort, light headedness or dizziness.
At the week 6 assessment, the programme will be reviewed and amended (where appropriate) according to participants’ functional/aerobic capacity and confidence (table 1 and online supplemental appendix 1).
Between visit 1 and visit 2
Weekly targets and progressions or regressions will be facilitated by supplementary telephone calls from week 0 to week 6.15 These will be delivered by the physiotherapist to provide remote monitoring and support regarding the HBEP. The focus will be to:
Assess compliance to the study intervention.
Discuss the severity of fatigue.
Discuss adverse effects that have not been reported in the interval prior to last communication with the patient.
Review of exercises to give patients an opportunity to discuss any concerns or ask questions related to the exercise programme.
Determine the need for progression or regression of the HBEP, including any individualised modifications that need to be made. For instance, modifications will be made if participants find a particular exercise too difficult (or easy) or are not reaching training zones of a self-reported RPE of 12–14. The activity time can also be increased and rest phase decreased, in the event participants report the programme to not be challenging enough (and vice versa).
Following the week 6 assessment (visit 2), telephone support will be withdrawn to assess long-term adherence to the study intervention.
Visit 2
At week 6 after the intervention, participants will be invited back to re-evaluate all baseline functional/aerobic capacity tests, HRQoL scores, and symptom severity and ensure appropriate adjustments to the HBEP are made.
Visit 3
The intervention will conclude at week 12, at which point investigators will assess all aforementioned parameters a final time. Participant diaries and accelerometers will be collected at this point.
Project objectives
Determine whether HBEP intervention is feasible and safe.
Identify whether HBEP attenuates fatigue in patients with PBC.
Outline the longevity in improvement in symptoms of fatigue and HRQoL once health-related telephone support is withdrawn.
Outcome measures
The overarching goal of this study is to determine the feasibility of HBEP among patients with PBC with clinically significant fatigue, with the decision to proceed to a subsequent RCT made on the following criteria:
Sample size: >25% of sample population (ie, patients with PBC and fatigue) eligible for study.
Patient consent: >66% of eligible study population consented to the study.
Safety: No severe adverse effects related to the study intervention.
Adherence: >80% adherence with HBEP for a minimum of 6 weeks (measured by self-reported diaries).
Our primary efficacy measure will be a reduction in median fatigue severity score according to the PBC40 QoL assessment tool. The PBC40 HRQoL assessment tool has been developed and extensively validated for use among patients with PBC, with the fatigue domain being the most commonly applied measure of treatment efficacy used in clinical trials.16–18
Secondary measures of efficacy will be gauged by changes in the following assessment tools, and justification for use can be found in online supplemental appendix 2:
FIS.19
The other domains of the PBC40 QoL tool, relating to cognitive, social, emotional, pruritus and overall symptoms.16
ISWT.23
SPPB.25
CLDQ.20
ESS.22
HADS.21
DASI.26
Liver biochemical parameters, namely, serum alkaline phosphatase, bilirubin and alanine transaminase.
GENEActiv physical activity and sleep quality.
Additional exploratory analysis will be conducted to study associations between fatigue and baseline patient demographics, seasonality and underlying UDCA response status, alongside the other domains of the PBC40 questionnaire.
Data capture
All qualitative and quantitative data will be entered into a purposely designed, secure access database and data subsequently analysed (SPSS V.24; IBM). Feasibility decision rules and clinical outcome measures will be presented using descriptive statistics. Quantitative changes before and after the intervention (from baseline to week 6 and to week 12) will be tested using a non-parametric statistical hypothesis for repeated measurements (Wilcoxon signed-rank test).
Adverse events
There will be a strict process for reporting adverse events (AEs) and adverse reactions (ARs), which will commence at screening and continue until the final participant has completed study intervention. All AEs, serious AEs (SAEs), ARs and serious unexpected ARs will be reported to the CI and sponsor’s research and development department via a dedicated case report form (CRF). Only AEs or SAEs that can be deemed probable or absolutely related to the study will be reported to the Research Ethics Committee.
Storage of data
All data relating to the study will be collected by the Co-I and recorded in standardised CRFs. Each participant will be given a unique study number at the time of consent and used as a way of identification. Data will be collected from the participant at point of entry into the study until completion of the intervention. All clinical data will be stored securely as per NHS regulations for a minimum of 15 years. All data documented on the CRF will be entered into an NHS password-secured computer and in concordance with the Data Protection Act 1998. All essential written documentation will be stored in line with the appropriate regulatory requirements and restricted to researchers essential to the study. Coded research data will be stored for 5 years anonymously under the property of the Queen Elizabeth Hospital UHB in keeping with good clinical practice.
Case report forms
CRFs will be completed at week 0 (baseline), week 6 and week 12 (end of study), and include all questionnaire and functional outcomes. Electronic CRFs will be completed on an NHS Trust password protected system and will include medical history, eligibility screening and study treatment adherence.
Sponsorship, indemnity and monitoring
UHB will act as sponsor for the duration of the study. As sponsor, UHB will be responsible for the conduct of the study and indemnify the study centre against any claims, arising from any negligent act or omission by the hospital in fulfilling the sponsor role in respect to the study. The study is supported by an unrestricted grant from Intercept Pharmaceuticals
Discussion
This is the first feasibility trial investigating HBEP in the treatment of refractory fatigue and QoL in patients with PBC. To date, 262 individuals with PBC have been preidentified, of whom 82 are deemed eligible, and 42 agreed to attend a dedicated screening visit.
Safety
Supervised exercise sessions and advice are well supported within chronic disease, but there are very few that focus on home-based exercise and how this influences fatigue. Moreover, there are a limited number of large RCTs that report safe use of exercise therapy in patients with chronic liver injury, most often among patient groups with advanced disease.
Combined with education and the chosen objective measures, participants will have clear training guidelines and supporting information along with contact numbers to reduce the risk of AEs. With supplementary weekly telephone calls across the first 6 weeks, the opportunity to discuss any concerns or AEs will be available.26 Rigorous exclusion criteria will help to reduce the risk of AEs and maximise the safety of remote exercise intervention that is monitored via health telephone calls. This will allow for maximal participation while ensuring a safe exercise regimen is followed.
The intervention itself has been developed and based on well-documented moderate-intensity training models and extrapolated from an exercise protocol, which demonstrated the safety of such a HBEP in patients with more advanced liver injury.15
Challenges to study design
One in 5000 individuals are affected by PBC, with >90% of those being women aged between 30 and 65 years.3 Consequently, many eligible participants are in some form of employment and have family responsibilities. This will need to be considered when recruiting to the study, and where possible, ensure that research-related visits link with clinic visits to reduce participant burden.
As the study intervention is predominantly home-based, education and support to aid adherence are fundamental. Although it is well recorded that debilitating fatigue and its impact on HRQoL is the most widely reported symptom of PBC, very little is understood about individual influences. To aid in the assessment of adherence, participants will be asked to complete a study diary, with the aim of tracking self-reported physical activity and providing aggregate data in relation to weekly targets. Participants will also receive written information and pictures of their individualised exercise programme, with an opportunity to record exercises digitally. The intervention itself has been developed and based on well-characterised moderate-intensity training models and extrapolated from an exercise protocol that demonstrated the safety of HBEP among patients with more advanced liver injury.15
As fatigue differs greatly between individuals, the study intervention needs to offer a variety of exercises that can be progressed and regressed while still improving functional/aerobic capacity. Multiple intensity levels offer a unique approach as have been designed to help ensure the interventions were individually targeted with the flexibility to adapt if needed.
Future RCT considerations
This phase 1, single-centred feasibility study is required not only to demonstrate the safety of the intervention but also to highlight recruitment, dropout rates and adherence. Without this information, it would not be possible to power the correct number of participants needed for future RCTs.
Summary
To the best of our knowledge this is the first study to investigate HBEP in patients with PBC with debilitating fatigue. Enrolment for participation in the study is completed, and final results are expected in early 2021.
Acknowledgments
We would like to thank the PBC Foundation, PSC Support, and the Birmingham Gastroenterology and Liver Patient and Public Involvement Group for their input and advice into designing the study protocol.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Twitter @alicefreer18, @liverphysio, @cholestasisdoc
PJT and MJA contributed equally.
Contributors AF, lead physiotherapist delivering intervention, wrote the first version of the manuscript and approved the final/revised version to submission. FW and SD developed the study protocol design, provided critical insight into manuscript content and approved the final/revised version to submission. JH helped with the study design and selection of eligibility criteria, provided critical insight into manuscript content and approved the final version to submission. PT and MA helped with the study concept and overall supervision of the project and provided critical insight into manuscript content, revisions and final manuscript approval. PT and MA are guarantors of the article.
Funding This work was supported by an unrestricted grant from Intercept Pharmaceuticals.
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval The National Research Ethics Service (NRES) Committee and Health Research Authority London—Riverside Research Ethics Committee (IRAS 253115) approved version 1.1 of the study protocol. All participants will provide informed written consent.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data sharing is not applicable as no data sets are generated and/or analysed for this study. Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Data sharing plan with regard to study findings will be submitted alongside full trial results.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.