Article Text

Abstract
Nivolumab is an immune checkpoint inhibitor used to treat multiple solid-organ malignancies. While many of its immune-related adverse events are well established, nivolumab-induced sclerosing cholangitis remains poorly characterised, with no defined diagnostic criteria. Moreover, data regarding long-term outcomes are particularly lacking. We present a biopsy-proven case of nivolumab-induced sclerosing cholangitis, which uniquely captures 18 months of follow-up post-treatment. Our case highlights key features of intrahepatic subtype sclerosing cholangitis and suggests durable response to corticosteroid therapy.
- cholestatic liver diseases
- cancer immunobiology
- liver function test
- drug toxicity
- autoimmune biliary disease
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
- cholestatic liver diseases
- cancer immunobiology
- liver function test
- drug toxicity
- autoimmune biliary disease
Introduction
Immune checkpoint inhibitor (ICI) therapies have revolutionised the management of multiple malignancies. ICIs act by blocking inhibitory receptors on cytotoxic T cells, thereby promoting T-cell activity and inhibiting immunotolerance to tumour cells.1 Programmed cell death 1 (PD-1) and PD-1 ligand 1 inhibitors have been shown to improve survival in both treatment-naïve and pretreated patients with non-small cell lung cancer (NSCLC).2 3 However, unleashing suppressed T-cells can also induce immune-related adverse events (irAEs).2 3 Although severe irAEs are uncommon, they can be profound, necessitating treatment with immunosuppression and cessation of the culprit ICI.
Nivolumab is an anti-PD-1 monoclonal antibody, which has demonstrated durable objective responses in patients with NSCLC.4 While the most frequently observed serious gastroenterological irAEs from nivolumab are colitis and hepatotoxicity, immune-mediated sclerosing cholangitis (SC) is seldom described.4 In a recent systematic review, Onoyama et al5 analysed 31 cases of PD-1 inhibitor-related SC and noted that no diagnostic criteria exist for this irAE. Furthermore, data remain especially limited regarding its prognosis post management.
We therefore present this case of nivolumab-induced SC, detailing novel long-term follow-up data postcorticosteroid treatment. The patient provided written informed consent.
Case report
A 79-year-old male current smoker with minimal ethanol intake and stage IV lung adenocarcinoma (epidermal growth factor receptor and anaplastic lymphoma kinase wild-type) was admitted with markedly cholestatic liver function tests (LFTs) 8 months postcommencing second-line nivolumab therapy.
Background
He had initially been treated with 4 cycles of carboplatin and gemcitabine, with no response. On progression, he was changed to fortnightly nivolumab (3 mg/kg). He achieved a near-complete response on CT after five cycles, which was durable. His only prior irAE was diarrhoea, which was common terminology criteria for adverse events V.5.0 grade one and successfully treated with 1 month of oral prednisolone (25 mg daily for 1 week, 12.5 mg daily for 3 weeks). Medical history included a left nephrectomy for a low-grade papillary carcinoma, chronic obstructive pulmonary disease, gout and osteoarthritis. There was no antecedent history of gallstones or inflammatory bowel disease (IBD). His medications included tiotropium, allopurinol and oxycontin. His Eastern Cooperative Oncology Group (ECOG) performance status was 2.
Presentation
His cholestatic LFTs were detected when he presented for cycle 17 of nivolumab. He reported 2 months of mild abdominal discomfort. He did not have any diarrhoea or symptoms of jaundice. On examination, his vital signs were stable. He was tender in the epigastrium, but had no signs of peritonism. His Murphy’s sign was negative.
Investigations
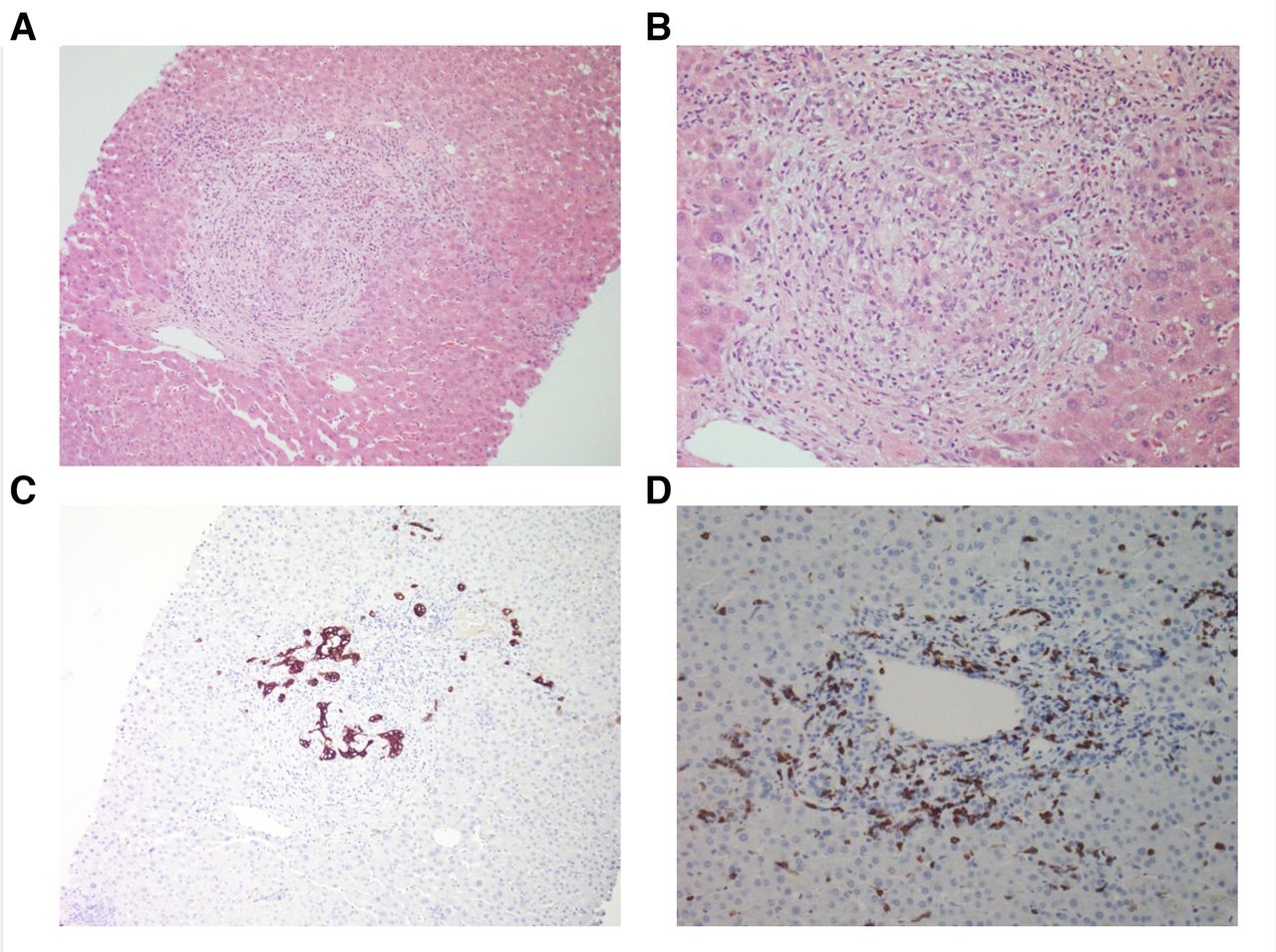
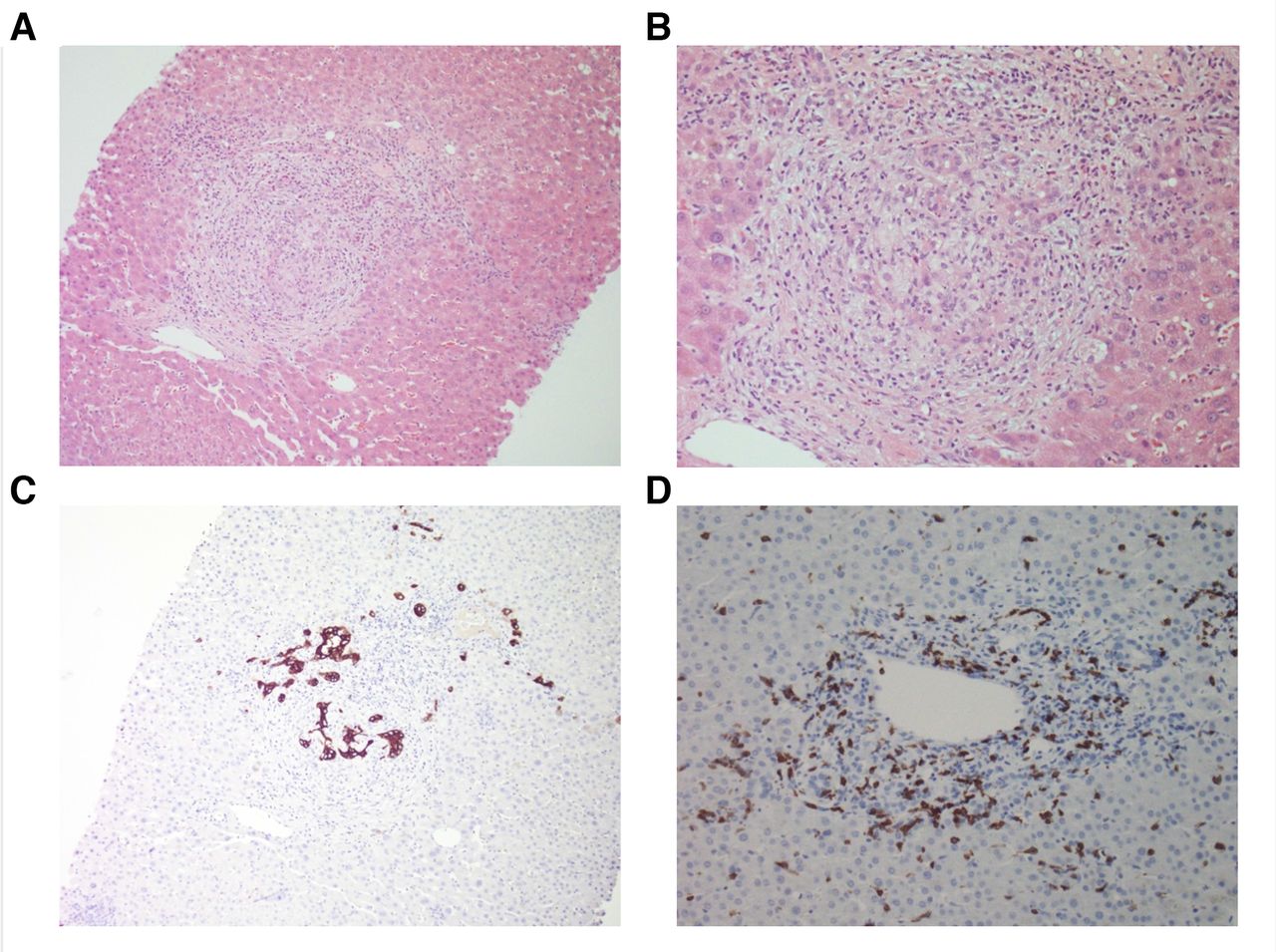
His LFTs (depicted in figure 1A) were normal at baseline, as was a previous liver ultrasound. At presentation, his alanine transaminase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP) and gamma-glutamyl transferase (GGT) were 156 (<40), 106 (<35), 1652 (<110) and 1249 (<60) U/L, respectively. Synthetic function (bilirubin, coagulation profile) was normal. Liver autoantibodies, IgG4 and faecal calprotectin were normal. MR cholangiopancreatography (MRCP) demonstrated ‘beading’ of the intrahepatic bile ducts (figure 1B). There was no evidence of any biliary hypertrophy, extrahepatic dilation or filling defects suggestive of gallstones. A liver biopsy demonstrated a destructive cholangitis and a portal inflammatory infiltrate with a mixed profile—including CD8+ T cells, histiocytes, neutrophils and eosinophils (figure 2).

A Liver enzymes (U/L) over time. * Nivolumab cycles 13–16, (1) admitted, (2) MRCP, (3) liver biopsy, (4) steroids. ALP, alkaline phosphatase; ALT, alanine transaminase; AST, aspartate aminotransferase; GGT, gamma-glutamyl transferase. B MRCP imaging which demonstrates intrahepatic beading.
{kind=link}
{kind=link}
Liver histopathology. (A) H&E at ×100 magnification, (B) H&E ×200, (C) Keratin 7 immunohistochemistry (IHC) ×100, (D) CD8+IHC ×200. (A) and (B) demonstrate portal stromal oedema with a mixed inflammatory infiltrate, destructive cholangitis and reactive bile ductules; (C) highlights florid bile duct injury, with remnants of damaged bile ducts among the portal infiltratel; (D) depicts the CD8+T cells in the portal infiltrate.
Treatment
A diagnosis of nivolumab-induced SC was made. Accordingly, nivolumab was ceased. Clinically, his abdominal discomfort resolved rapidly over 2 days. His ALT, AST and ALP improved, however, his GGT remained ≥1046 U/L. Therefore, a decision was made to commence 10 weeks of oral prednisolone (50 mg daily, weaned by 5 mg per week), during which his GGT reduced by >60% to 408 U/L.
Outcomes
After 18 months of follow-up, the patient remained well and ECOG 2. His ALT and AST were normal, and his ALP and GGT had nadired at 147 and 200 U/L, respectively. Despite ceasing nivolumab, his NSCLC remained stable on CT staging — without any subsequent therapy. A subsequent MRCP for research purposes was not performed due to COVID-19 restrictions.
Discussion
This case demonstrates the long-term outcome of nivolumab-induced SC postcorticosteroid treatment, and is of high clinical relevance as it uniquely captures 18 months of follow-up.
Nivolumab-induced SC is rarely diagnosed, with few cases in the literature. Our case highlights key clinical and biochemical findings consistent with those seen in other studies. Onoyama et al5 found that PD-1 inhibitor-related SC was most frequently reported in patients with the following demographics: male (68%), with NSCLC (62%), receiving nivolumab (61%). As with our case, patients commonly had abdominal pain (11/31, 35%), cholestatic LFTs without hyperbilirubinemia (median ALT 125 U/L, AST 129 U/L, ALP 1543 U/L, GGT 452 U/L; bilirubin 12.86 umol/L) and a normal IgG4 (12/13 tested, 92%).5 While the median number of cycles until onset of SC in others was 5.5, the 16 cycles seen in our patient falls within the observed range (1-27).5
In our case, a multidisciplinary pathological evaluation was key to diagnosing this irAE. Yet throughout the literature, diagnostic approaches have varied considerably. Onoyama et al5 noted that most cases had radiological abnormalities, such as biliary dilation (20/26, 76.9%), hypertrophy (20/21, 95.2%) or stenosis (8/23, 34.8%). As with other studies,6 cases were categorised into novel subtypes: ‘extrahepatic’, with diffuse extrahepatic hypertrophy without stensosis (15/31, 48%); ‘diffuse’, with multiple extrahepatic and intrahepatic biliary stenoses (4/31, 13%); and ‘intrahepatic’, with multiple intrahepatic stenoses (3/31, 10%)5—as seen in our patient. However, less than half of cases were biopsy proven (15/31, 48.4%). Of the histological changes observed, most had a biliary and/or periductular inflammatory infiltrate (14/15, 93.33%) of predominately CD8+ T cells (8/15, 53.33%). These features were present in our case and are in keeping with the mechanism of ICI action.5
The establishment of diagnostic criteria for nivolumab-induced SC is critical. Although rare, this irAE can cause significant bile duct injury and, as our case demonstrates, ICI cessation and treatment can result in clinical and biochemical improvement. Kawakami et al7 previously suggested that nivolumab-induced SC could be characterised by 6 features: (1) cholestatic LFTs, (2) normal/low autoantibodies and IgG4, (3) localised extrahepatic biliary dilation without obstruction, (4) diffuse extrahepatic biliary hypertrophy, (5) CD8+ T cell biliary infiltrate on liver biopsy and (6) partial/no improvement of LFTs with corticosteroids. However, as highlighted in our case, not all of these features are always present in PD-1 inhibitor-related SC.5 Moreover, this diagnostic approach would effectively miss any ‘intrahepatic’ or steroid-responsive subtypes.
In our patient, corticosteroid therapy temporally correlated with a substantial improvement in the GGT. In the literature, a variety of treatments have been used for PD-1 inhibitor-related SC, with varying efficacy. These include: corticosteroids (26/31, 83.87%), other immunosuppressants (mycophenolate mofetil 6/31, 19.35%; tacrolimus 1/31, 3.23%), primary biliary cholangitis (PBC) therapies (ursodeoxycholic acid 13/31, 41.94%; bezafibrate 1/31, 3.23%) and biliary drainage (6/31, 19.35%).5 Of the cases treated with corticosteroids, most demonstrated some LFT improvement (15/26, 57.70).5 However, very few achieved biochemical normalisation (3/26, 11.54%) and in some cases LFTs did not improve or even worsened (8/26, 30.77%).5 Therefore, Onoyama et al5 did not recommend steroid therapy for PD-1 inhibitor-related SC,5 which contrasts the general approach to treating irAEs outlined in international guidelines.8
The goals of steroid therapy for immune-mediated SC remain unclear, however, as most patients do not normalise their LFTs.5 As we uniquely demonstrate, our patient remained well and high functioning over 18 months of follow-up, despite some persistent biochemical abnormalities. Moreover, it has been suggested that steroid-responsiveness may depend on the subtype of PD-1 inhibitor-related SC.5 While the numbers are small and the concept novel, Onoyama et al5 recognised that the intrahepatic cases treated with corticosteroids responded (2/2 LFTs improved or normalised, 100%), as opposed to the extrahepatic or diffuse cases (1/15 LFTs normalised, 6.67%). Ultimately, further data are required to inform diagnostic and treatment decision making in this field.
The pathogenesis of this irAE also remains unclear. Interestingly, even in well-established cholangiopathies such as PBC and primary sclerosing cholangitis (PSC), the mechanisms leading to bile duct injury are incompletely understood. Immunological factors have been implicated in both conditions, as PBC is closely associated with the anti-mitochondrial autoantibody and PSC with IBD.9 10 Moreover, a T-cell response to bile duct epithelial cells is a key feature of PBC,9 that has also been suggested in PSC.11 Potential culprit biliary antigens include molecules with epitopic similarity to a mitochondrial enzyme in PBC,12 or to colonic epithelial cells in PSC.13 Whether these antigens cross-react with tumour molecules remains unclear. Other potential mechanisms underlying cholangiopathies include genetic susceptibility, biliary acid retention or bacterial factors such as portal circulation permeability, duodenobiliary reflux or microbiomial changes.10 14 15 Additional research into whether these factors contribute to PD-1 inhibitor-related SC could assist in explaining why differing subtypes of varying steroid-responsiveness occur, and help clinicians to identify patients at risk of this irAE.
Conclusion
Nivolumab-induced SC is an uncommon diagnosis, but may occur at any time during ICI therapy. It should be considered in patients with abdominal discomfort, cholestatic LFTs and radiological biliary abnormalities, and confirmed via histological diagnosis. Our data support the use of steroids in patients with intrahepatic subtype SC, consistent with prior studies. Long-term, we uniquely highlight that clinical outcomes in patients whose cholestasis improves with steroids are excellent, and that a durable response is maintained after steroid cessation, even if LFTs do not completely normalise. Ultimately, further studies are needed to better understand both why and how select patients develop this rare toxicity.
References
Footnotes
Contributors All of the authors made substantial contributions to the conception or design of the work, or the acquisition, analysis or interpretation of data, drafted the work and revised it critically, approved the final approval of the version submitted, agree to be accountable for all aspects of the work.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests TJ reports personal fees from AstraZeneca, personal fees from Bristol Myers Squibb, personal fees from Roche, personal fees from Pfizer, personal fees from Novartis, personal fees from Specialised Therapeutics, personal fees from Amgen, personal fees from MSD, personal fees from Merck and personal fees from Takeda, outside the submitted work.
Patient consent for publication Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available on reasonable request. Further deidentified patient data are available on request.