Article Text

Abstract
Objective Pelvic radiotherapy is used to treat 17 000 people in the UK each year. Eight in 10 develop difficult bowel problems during pelvic treatment, especially diarrhoea, urgency and incontinence. Some cannot complete treatment, reducing the chance of cancer cure. Undertaking gastroenterologist-led investigation and management during pelvic radiotherapy has never been evaluated. In this study, we aimed to assess whether patients could successfully receive a novel gastrointestinal (GI) care bundle during chemoradiotherapy (feasibility aim) and would experience reduced symptom severity (clinical impact aim).
Design This randomised controlled trial recruited patients with cervical and bladder cancers undergoing radical chemoradiotherapy. Participants were randomised to intervention or control groups. Questionnaire and anthropometric data were collected. All intervention group patients received individualised dietary counselling weekly throughout treatment, and if bowel symptoms developed they were offered rapid-access investigation and treatment for any identified pathology: lactose intolerance, bacterial overgrowth or bile acid malabsorption.
Results Feasibility: 50 participants were recruited, 24 were randomised to the intervention group and 26 to the control group. All completed 20 fractions of external beam pelvic radiotherapy. It was possible to perform 57/72 (79%) of proposed intervention tests with no disruption of oncological management.
Clinical impact All participants developed GI symptoms during radiotherapy. The median symptom score for each group increased from baseline at 6 weeks. This was from 0.156 (0.000–0.333) to 0.600 (0.250–1.286) in the control group, and from 0.00 (0.000–0.300) to 0.402 (0.000–0.667) in the intervention group.
Conclusion It was feasible to recruit to and deliver a randomised controlled trial of interventions in patients undergoing pelvic chemoradiotherapy. Lower median bowel scores were reported in the intervention group at 6 weeks, with fewer patients experiencing symptoms overall.
Trial registration number ISRCTN783488.
- radiotherapy
- clinical trials
- radiation enteritis
- cancer
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Summary box
What is already known about this subject?
Pelvic radiotherapy causes significant gastrointestinal toxicity that can limit the effectiveness of treatment or even curtail it. The acute effects on the gastrointestinal tract may be amenable to intervention if identified and treated. However, gastroenterologists are rarely involved in clinical management.
What are the new findings?
In this preliminary feasibility study a bundle of investigations and interventions was possible and acceptable to patients with no disruption to cancer treatment.
How might it impact on clinical practice in the foreseeable future?
A larger fully powered study is now required to study the clinical impact of intervention on symptoms and outcomes.
Introduction
As treatments improve, more people are surviving cancer.1 In the UK 17 000 people undergo pelvic radiotherapy per year.2 Although much attention has focused on the devastating late effects of treatment (pelvic radiation disease), there has been less focus on symptoms arising during radiotherapy. Patients almost universally experience acute lower gastrointestinal (LGI) symptoms.3 If symptoms are severe treatment may need to be interrupted or stopped with a resulting decreased chance of cure.4 It is also reported that patients who experience more severe acute bowel effects or more prolonged GI symptoms during radiotherapy have an increased risk of experiencing late bowel effects.5 6 It is therefore hypothesised that amelioration of acute toxicity, as well as improving quality of life (QOL) and success of oncological therapy, could reduce the likelihood of developing late effects in the GI tract.7
As yet there is a paucity of evidence for interventions which decrease acute GI symptoms seen with pelvic radiotherapy,8 so management is empirical and symptomatic, and usual practice does not involve the approaches that would be taken by gastroenterologists to such symptoms in other clinical contexts. When patients report diarrhoea they are generally simply advised to adhere to a low-fibre diet but evidence for this intervention is lacking,9 and they may be only treated with antidiarrhoeal agents.
In planning this project it was clear that more specific and tractable pathophysiological entities such as small intestinal bacterial overgrowth (SIBO), bile acid malabsorption (BAM) or lactose intolerance (LI) would not generally be considered in a pure oncology environment, potentially excluding patients from clinically useful interventions. During pelvic radiotherapy treatment these specific small bowel pathologies occur in 26%,10 44%–57%11 12 and 15%–44%,10 11 of patients, respectively. However, the evidence for interventions in this clinical setting is currently lacking. There is also a prevalence of 11%–33% malnutrition in patients prior to receiving pelvic radiotherapy.13 Undernutrition and low body mass index (BMI) are known to increase the risk of acute and late GI toxicity.14 15 A Cochrane review found a positive effect of several different dietary interventions compared with control in reducing the severity of diarrhoea during pelvic radiotherapy.16 A randomised controlled trial (RCT) was performed in patients undergoing neoadjuvant radiotherapy for colorectal cancer.17 They included three arms: a control group, a group who received nutritional supplements and a group who received individualised nutritional counselling during their radiotherapy treatment. The third group maintained their intake and demonstrated less nutritional status decline during therapy and had fewer and less severe late effects, improved quality-of-life and survival compared with the other two groups. These data are supportive of the positive effects of nutritional intervention on several important outcomes.
Therefore we aimed to undertake the first reported study of a targeted GI care bundle combining nutritional and clinical interventions in the management of patients while concurrently undergoing pelvic chemoradiotherapy. This study was a RCT in patients with cervical and bladder cancers who were undergoing curative chemoradiotherapy.
The central aim of study design and delivery was to test the feasibility of recruitment, randomisation, investigation, treatment and follow-up assessments in patients developing acute GI symptoms during radiotherapy. This is an onerous phase for patients, and it is important to understand whether additional investigation and intervention would be acceptable and deliverable during intercurrent therapy and its associated toxicity.
The primary clinical endpoint was severity of GI symptoms immediately post-treatment as determined by maximum and average score from the Common Toxicity Criteria for Adverse Events (CTCAEs) bowel subset (week 6) to determine if a GI care bundle, including nutritional intervention and detecting and treating LI, SIBO and BAM, improves GI symptoms in the short-term.
The study was primarily funded and essentially designed to test the feasibility of collecting data including patient-reported outcome measures to inform a larger definitive study of effectiveness and cost effectiveness if a subsequent multicentre RCT were to be undertaken.
Secondary aims of the project were to determine whether this intervention reduced treatment interruptions and enabled more patients to complete chemoradiotherapy by reducing the severity of acute toxicity; improved QOL during and after treatment; and reduced the frequency of late effects.
Methods
Unselected patients with pelvic (cervical and bladder) cancer who were to undergo chemotherapy and radical pelvic radiotherapy were recruited.
To be eligible, patients had to be over 18 years of age and have a diagnosis of histologically confirmed cervical or bladder cancer. The two pathologies were not stratified in the design. Exclusion criteria included pre-existing bowel disease, inability to complete patient-reported questionnaires and inability to give consent. Eligible patients were given a study patient information leaflet and were given at least 24 hours to consider the study information and an opportunity to ask questions prior to consenting to the study. On or before their first day of treatment patients were consented to the study by one of the coinvestigators. They were then randomised to ‘Treatment as usual’ (TAU) or ‘GI Care Bundle’ by the Christie Hospital Clinical Trials Unit randomisation telephone line. Clinical Trials Unit staff had no access to future allocations. The randomisation process was a computer-generated random sequence on a 1:1 basis in a single block of 60. It was evidently not possible to blind this study due to the nature of the intervention, though data analysis was fully blinded.
Questionnaire and anthropometric data were collected. The CTCAE is the only system which is designed to collect data on acute toxicity and late effects for all cancer treatments.2 The Late Effects on Normal Tissues–Subjective, Objective, Management and Analytic system was developed into questionnaires which include physician and patient-recorded data on toxicity.18 These questionnaires have been used at this centre to collect toxicity data from hundreds of gynaecological patients undergoing pelvic radiotherapy. This validated patient-reported questionnaire is designed to detect side effects of pelvic cancer treatments comprising bowel, bladder and sexual domains which detect symptoms and their severity.19 A mean score for each of the three domains between 0 and 4 is calculated. The higher the derived score, the greater the symptom severity for each domain. Where possible the 12-week and 12-month assessments were timed to fit in with routine oncology follow-up visits. Anthropometric measurements included height measured using a portable stadiometer to the nearest 0.1 cm, weight measured to the nearest 0.1 kg on calibrated scales (Seca 875 flat scale), BMI, and handgrip strength using the non-dominant hand (Takei 5101 Grip Dynamometer Analogue). Bioelectrical impedance was measured (Bodystat 1500 machine, Isle of Man, Bodystat Ltd) with the patient in a supine position.
The intervention involved individualised dietary counselling by the study dietician using the anthropometric data, patient-reported subjective, global assessment score, malnutrition universal screening tool score, dietician symptom assessment and 3-day food diary to tailor dietary advice and prescription of oral nutritional supplements where necessary.
If the participants in the intervention group reported the development of any LGI symptoms (at trial visit or to radiographers or clinicians while attending for treatment), three investigations were arranged as a bundle of tests depending on how well the patient was feeling and their treatment schedule. While awaiting study tests, intervention group participants were given symptomatic treatment for example, loperamide or Fybogel. The tests were a Selenium-75-homocholic acid taurine (SeHCAT) scan for BAM, a glucose hydrogen methane breath test for SIBO), and a lactose hydrogen methane breath test for LI. A rise in hydrogen of 20 ppm or a rise in methane of 15 ppm above baseline reading at any point during the test after wither sugar load was deemed a positive result. Patients with SIBO were treated with a 7-day course of ciprofloxacin (500–750 mg two times per day). If the patient was allergic to ciprofloxacin, had any contraindications or side effects or if there was no symptomatic response to ciprofloxacin, they would receive a 7-day course of doxycycline (200 mg once daily on day 1, then 100 mg daily thereafter). If the patient was allergic to doxycycline, had any contraindications or side effects or if there was no symptomatic response to doxycycline, they would receive a 7-day course of clarithromycin (250–500 mg two times per day). Patients with BAM (<15% retention at 7 days) were treated with colestyramine 1 to 3 sachets daily titrated to symptoms. If this was not tolerated by the patient, they were treated with colesevelam (625 mg 1–7 times daily). All patients were advised to take bile acid sequestrants at least 1 hour after other medications. None of the patients were taking oral chemotherapy.
If LI was identified, patients were counselled by the dietician regarding a low lactose diet.
The feasibility of collecting patient-reported outcome measures and resource data for an economic evaluation within a future trial was also tested. Health-related quality-of-life (HRQL) as measured by the EQ-5D-5L was collected at baseline, 6-week, 12-week and 12-month follow-up.
This feasibility study aimed to collect initial data for the primary outcome measures. The following results are primarily descriptive in nature. For the purpose of a pilot study recruitment and retention of at least 30 patients per arm is deemed sufficient to estimate key parameters with sufficient precision that they may be used to inform the calculation of power for a larger trial.20
The study was completed at the Christie NHS Foundation Trust, Manchester, United Kingdom supported by the Clinical Trials Unit. Adverse events were recorded and any serious adverse events relating to the trial intervention would have been reported to the clinical trials unit. Studies were conducted in accordance with the Declaration of Helsinki.
Data were analysed with IBM SPSS Statistics V.22 and Stata V.13.
Results
Fifty patients were recruited. The baseline characteristics of the intervention group and control group are shown in table 1.
Baseline characteristics of the intention-to-treat population
Of 117 eligible patients, 54 patients declined to participate and recruitment of 13 patients was not pursued (due to reasons such as distress relating to cancer diagnosis). Recruitment rate was 42.7%. Thirty of those who declined felt that participation in the study would be too much on top of standard treatment. Other reasons given for declining included child care commitments and a desire not to undergo extra tests.
Twenty-four patients were randomised to the intervention group and 26 to the control group. All patients remained in the study at the 6-week time point and were therefore assessed for the primary outcome. The trial Consolidated Standards of Reporting Trials flow diagram is shown in figure 1.

CONSORT flow diagram demonstrating recruitment and follow-up. CONSORT, Consolidated Standards of Reporting Trials.
The patients with bladder cancer received weekly gemcitabine chemotherapy and the patients with cervical cancer received weekly cisplatin chemotherapy while undergoing 4 weeks of radiotherapy treatment. All trial patients received 20 fractions of external beam radiotherapy as planned. The patients with bladder cancer received 3D conformal radiotherapy (3D CRT) and the patients with cervix cancer received 3D CRT or volumetric modulated arc therapy. For further details of planned and delivered treatment see table 2. There were no radiotherapy interruptions due to treatment toxicity. The mean treatment volume of the control group was 1148 cm3 versus a mean treatment volume of 845 cm3 in the intervention group (p=0.02). The planned treatment dose was 40 Gy for the patients with cervix cancer and 52.5 Gy for the patients with bladder cancer.
Planned and delivered treatment
Brachytherapy was originally planned for 13 control patients of whom 10 received this as well as a further 2 patients who were not originally going to receive it. The patients who did not receive brachytherapy received an external beam boost. This was due to the large size or positioning of the tumour in these patients. At the start of treatment, 11 intervention patients were due to have brachytherapy and 9 received this. The other two intervention patients received an external beam boost. In both of these patients there was a failed attempt at brachytherapy. Of the patients in each group who received brachytherapy they all completed their planned treatment schedule. One patient in the control group had a 1 week delay of their first brachytherapy treatment due to a diarrhoeal illness which other members of her family had suffered from. All patients who underwent an external beam boost completed their planned 10 fractions of treatment with no treatment breaks or delays.
All patients in each group experienced LGI symptoms. There were no radiotherapy treatment interruptions and no physician-reported grade 3–4 bowel toxicity was recorded in either group. All trial patients completed 20 fractions of external beam pelvic radiotherapy. In the intervention group 17 (71%)/24 patients received the planned four cycles of chemotherapy versus 16 (62%)/26 patients in the control group. The reasons for this are detailed in table 3.
Reasons why less than four cycles of chemotherapy were delivered
Chemotherapy regimens were interrupted due to bowel symptoms in three patients in the control group and one patient in the intervention group. There were no serious adverse events attributable to the trial intervention.
All patients reported the development of GI symptoms within the first 6 weeks of radiotherapy treatment. The median bowel score of each group increased at 6 weeks and decreased at 12 weeks (table 4). However at 52 weeks the median bowel score increased compared with 12 weeks. A number of extreme scores were observed in both groups postbaseline. The control group score was higher than the intervention group score at 6 weeks (the primary endpoint). The 95% CI for the median difference (TAU−GI Care Bundle) was estimated to be (−0.05, 0.60).
The median bowel symptoms score of each group at each time point
There were a higher number of patients with a GI symptom score of 0 in the intervention group (8) versus the control group (only 1) at 6 weeks.
There was no clinical difference between the control and intervention group scores at any time point for anxiety, depression or total Health Anxiety and Depression Scale (HADS) score. There did not appear to be a clinical difference between the EORTC QLQ C30 scale scores between the groups. The individual and overall QOL scale scores were seen to deteriorate at the end of radiotherapy with subsequent improvement in both groups.
HRQL scores from the EQ-5D-5L were available for a high proportion of each trial arm (87.5% to 100%) at every follow-up time point. In both arms of the trial mean HRQL fell at the 6-week time point and improved slightly at the 12-week time point. Overall mean HRQL was at its lowest at the 52-week time point in both arms. The mean HRQL in the control group fell by 0.14 (0.91 (range 0.64–1)) at baseline to 0.77 (0–1) at 52 weeks compared with 0.06 in the intervention group (0.89 (0.53–1)) at baseline to 0.83 (0–1) at 52 weeks (figure 2). This relative difference was in part driven by the increased mortality observed in the control arm.
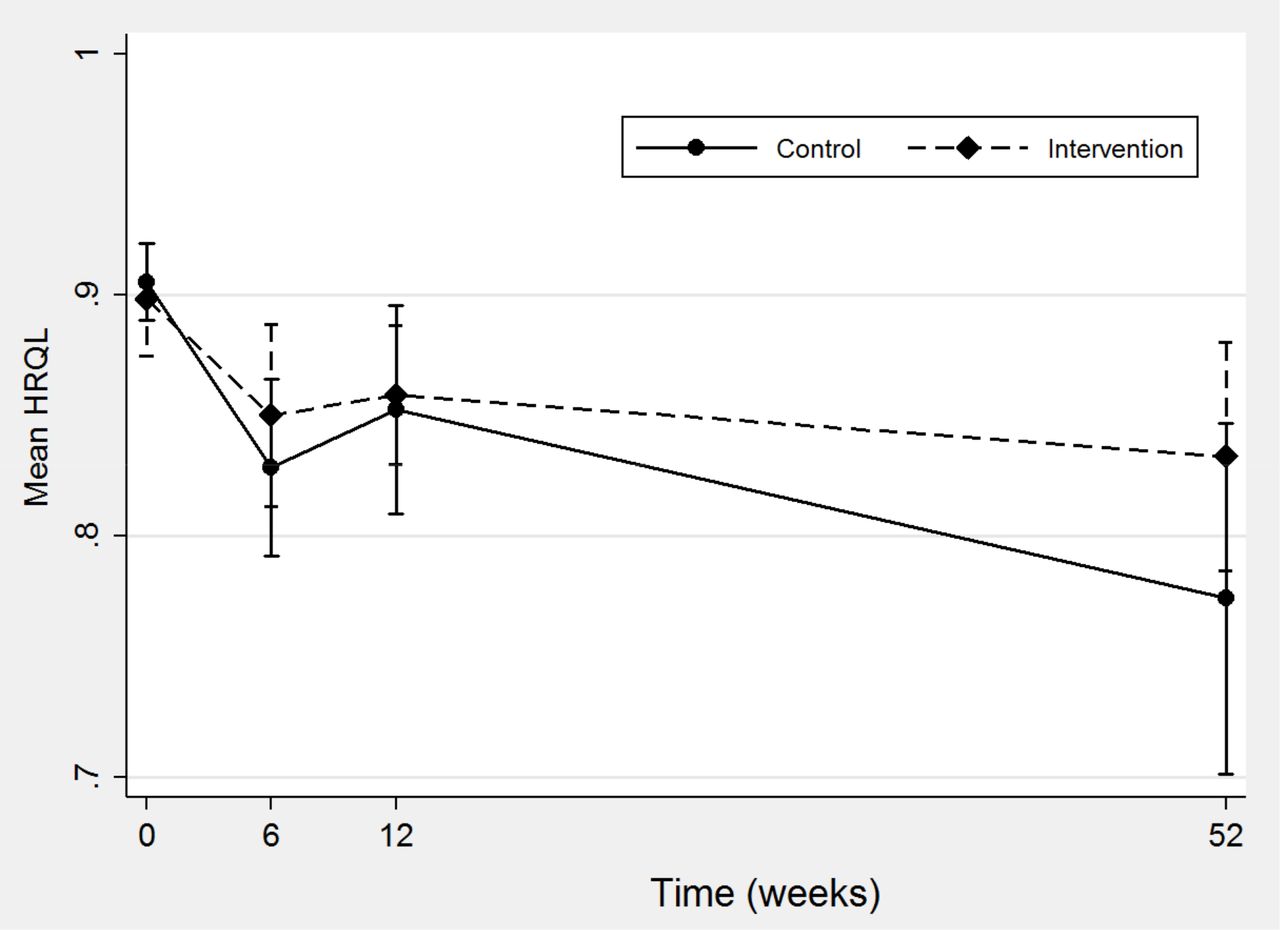
HRQL in control and intervention groups over the 52-week period of study. HRQL, health-related quality of life.
There was excellent compliance with questionnaires, summarised in table 5.
Questionnaire returns at each time point
No patients were underweight at baseline. Seventy per cent of the control group were overweight or obese at baseline compared with 54% of the intervention group. The collection of anthropometric data was feasible in both groups and throughout the study. These data are summarised in table 6.
Anthropometric measurements
All patients developed LGI symptoms during the first 6 weeks of the trial, therefore all intervention patients received some elements of the GI care bundle. All enrolled patients in the intervention arm were reviewed by the dietician at the specified time points as per protocol. Sixteen patients completed food diaries at all time points. Seven patients completed a proportion of the requested food diaries and one patient did not complete any food diaries. One hundred and fifty one (84%) of a requested 179 food diaries were completed. There was documented compliance with the oral nutritional supplement in 22/33 weeks leading to a compliance level of 67%.
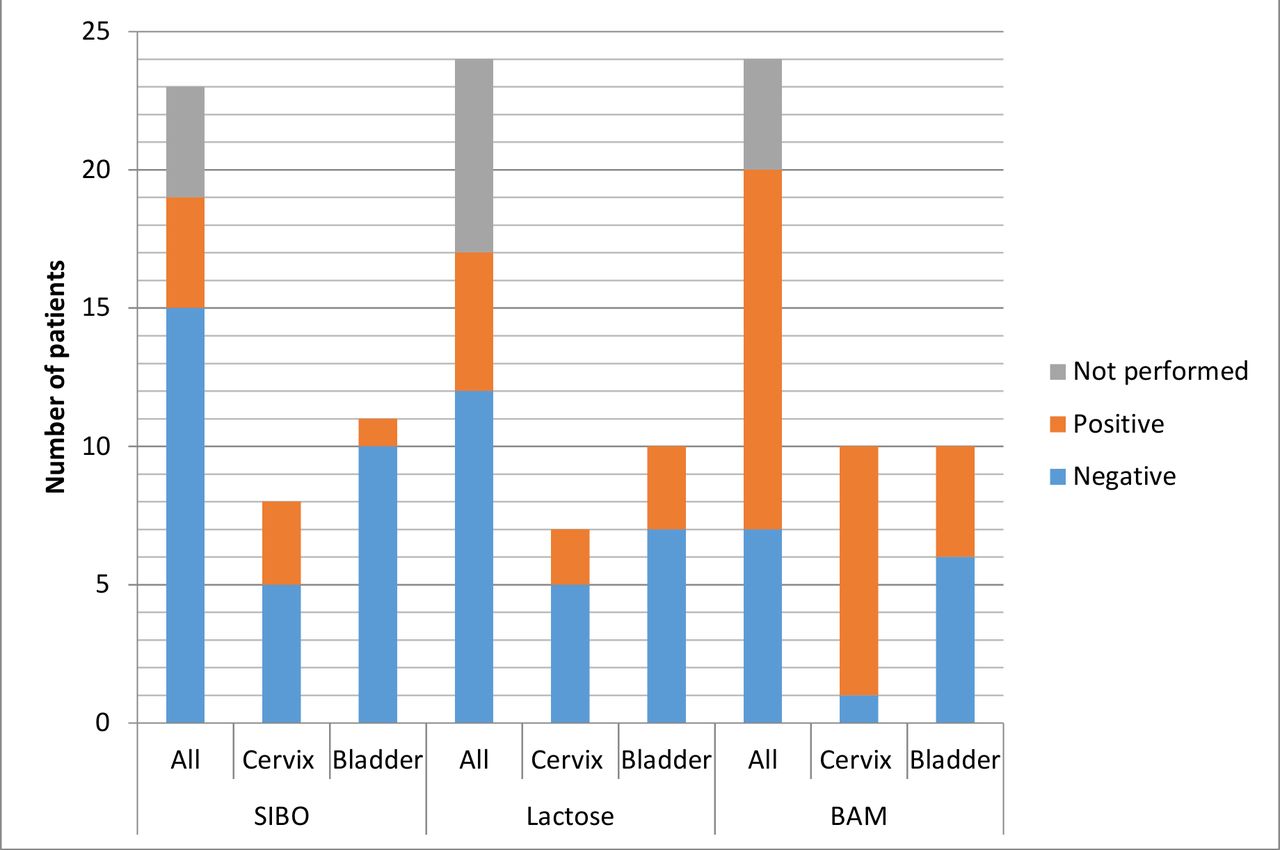
As all patients developed GI symptoms a total of 72 intervention tests should have been performed. It was only possible to perform 57 (79%) tests. Seventeen (71%) of the 24 intervention patients had all three tests. Reasons why patients did not undergo tests included intercurrent illness (six tests) and patient choice (9 tests in four patients). Figure 3 demonstrates the proportion of patients who tested positive for each diagnosis. One patient tested positive for all three diagnoses, and six patients tested positive for two diagnoses. This is detailed in table 7.

{kind=link}
{kind=link}
{kind=link}
Number of patients with positive and negative intervention test results and those which were recommended but not performed. Results shown for all intervention patients and also grouped by those with cervical cancer and those with bladder cancer. One SIBO breath test was inconclusive. BAM, bile acid malabsorption; SIBO, small intestinal bacterial overgrowth.
Results of intervention tests performed by study number and tumour type
The reasons why individual patients did not undergo all the proposed investigations are outlined in table 8.
Patients who did not undergo three intervention tests and reasons why tests were not performed
Finally, patient satisfaction was measured and it indicated excellent acceptability of the intervention. Irrespective of trial arm or time point, patient ratings of overall satisfaction were all either ‘Very Satisfied’ or ‘Extremely Satisfied’.
Discussion
Until now, there is limited evidence about the practicality and impact of interventions to improve gut symptoms and nutrition in patients undergoing chemoradiotherapy for pelvic cancer. It was feasible, and acceptable to patients, to recruit and deliver a RCT of GI intervention in patients undergoing curative chemoradiotherapy for cervix and bladder tumours. Furthermore there were no adverse events attributable to the intervention, and patient satisfaction with the trial was very high. There were 10 withdrawals in total.
All patients in both groups completed their planned external beam radiotherapy. The lack of radiotherapy treatment interruptions may be a reflection of modern radiotherapy techniques and patient selection. Patients who are thought to be at higher risk of bowel toxicity are often treated with radiotherapy only. Chemotherapy was halted in four patients due to bowel symptoms, allowing patients to continue with their radiotherapy treatment. Despite recent advancements in radiotherapy techniques this study found a comparable rate of BAM, LI and SIBO to that in previous studies.10–12
As anticipated there was an increase in GI symptoms and deterioration in QOL indices at 6 weeks in all patients in both groups suggesting significant symptom burden at this time point. This is in keeping with previous data regarding GI symptoms and QOL secondary to pelvic radiotherapy.19 21 Calculation of a 95% CI for the median difference in bowel score was suggestive of a lower median bowel score in the intervention group at 6 weeks. This now needs testing in a fully powered multicentre RCT. It is potentially noteworthy that eight patients had a 0 symptom score at 6 weeks in the intervention group, versus only one in the control group.
The relative completeness of HRQL data in this sample suggests that participants were not overly burdened with reporting tasks. The EQ-5D-5L does appear to be sensitive to changes in these participants self-reported health status. There is a marked decrease in HRQL in both arms at the 6-week follow-up, predictably reflecting negative short-term effects of chemoradiotherapy treatment. Acknowledging the small numbers of participants in this pilot study the EQ-5D-5L does, however, demonstrate the potential to differentiate between arms in terms of patient-level outcomes. Finally although there was a perceived difference between the arms in HRQL at 12 months this must be interpreted with caution due to the increased mortality in the control group.
The control group had a higher proportion of patients with cervical cancer. Although studies often group together patients with different pelvic cancers, this may not be appropriately exemplified by the difference in mortality found in this study. Furthermore the radiotherapy fields delivered will be different for different tumour types, different chemotherapy agents are given and there may be different pathophysiological processes underlying bowel toxicity. In this study 9/10 of patients treated for cervical cancer were found to have BAM, while only 4/10 patients treated for bladder cancer had BAM. Future trials should consider randomisation stratified by tumour type.
A recent Cochrane review found that dietary modification during pelvic radiotherapy may offer protection against diarrhoea.16 Studies of treatment of individual small bowel pathologies such as LI and BAM have not shown benefit.22 23 This study demonstrates that an individualised approach with testing of the small bowel and dietary counselling is possible and may show benefit of this strategy at 6 weeks. A smaller dataset was available at the 1 year time point and results at this time point should be interpreted with caution.
This study had several strengths. Previous studies of individual interventions for GI symptoms secondary to pelvic radiotherapy have struggled to show benefit. This study delivered a care bundle with tests to detect small bowel disease and dietetic input to ensure that patients already at risk of malnourishment received sufficient calories. A further strength of this study was the measurement of compliance. As this was a feasibility study it was important to measure compliance with the visits, questionnaires and anthropometric measurements, which was high in each instance. Compliance with dietetic input was high as shown by the return of food diaries and oral nutritional supplement sheets. As expected not all patients were able to undergo all intervention tests often due to the burden of their chemoradiotherapy regime or intercurrent illness. However, a pragmatic approach could be taken in a larger trial and in particular if patients are unable to undergo the SeHCAT scan then testing of C4 or FGF 1924 could be performed or a trial of colesevelam could be given.
Although all satisfied the recruitment criteria for a pelvic cancer and therefore all pathologies are equally eligible for the purposes of the study, the intervention and control arms were, despite random allocation, different in terms of age and primary tumour site, and therefore also in gender as all patients with cervical cancer are females. Therefore stratification by these factors at randomisation should be considered in a future trial.20 The recruitment rate of 42.7% likely reflects the timing of recruitment which by necessity occurs soon after patients receive a cancer diagnosis, which they are likely to still be processing. Due to this recruitment rate a sample size of 300 patients is required for a future multicentre trial.
Due to the nature of the intervention it was not possible to blind this RCT. The numbers in each arm compare favourably with other studies examining nutritional intervention in this patient group,25 26 despite this study primarily being a pilot study. One of the other main limitations of the trial was the unavoidable lack of baseline data regarding the presence of small bowel diagnoses in the intervention group. This would be something to consider including in the larger future trial to ensure that any positive tests performed during radiotherapy represented an effect of treatment on the small bowel. However, this has to be balanced with the potential delay to commencing chemoradiotherapy (which would not be appropriate) and burden on patients at a very difficult period. With reference to BAM, specifically it may be appropriate to measure FGF 19 or C4 prior to radiotherapy to prospectively identify and exclude patients with undiagnosed bile acid diarrhoea.24 A further limitation may be the choice of the 6-week time point to measure the primary outcome. This study visit was poorly attended, likely due to fatigue following treatment. One possible solution may be to widen the time point of the 6-week visit to plus or minus 2 weeks. Alternatively weekly scores could be summated to give an overall cumulative toxicity score for the acute toxicity period.
This pilot study demonstrated that it is feasible and acceptable to collect questionnaire, anthropometric and resource use data and deliver this bundle of care to patients undergoing radical chemoradiotherapy, and suggested that some benefit may be obtained at 6 weeks, a key time point in this treatment pathway. A further multicentre RCT of this intervention with embedded economic evaluation should be performed to confirm benefit of the intervention in terms of patient-reported acute GI toxicity and late GI effects. This is planned.
References
Footnotes
KLW and CCH are joint first authors.
KLW and CCH contributed equally.
Contributors CCH initially designed the study with input from SED, JTM, SL, MH and STB and initiated the study. KLW recruited to and ran the study to completion, cosupervised by SED and JTM, and wrote the first draft of the report with input from all authors. MH undertook the statistical analysis. ME analysed the HRQL data. CCH and KLW contributed equally to the study.
Funding This study was funded by the UK National Institute of Health Research’s Research for Patient Benefit Programme (reference PB-PG-0211-10024).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval Local governance approval was gained and ethical approval was obtained from National Research Ethics Services Committee (REC number 12/NW/0160).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. Via corresponding author to team ORCID: 0000-0001-6158-5135.