Article Text

Abstract
Objective To provide an overview of the pathology and molecular pathogenesis of traditional serrated adenomas (TSA).
Design Describe the morphology and molecules that play a role in their pathogenesis.
Results These exuberant polypoid lesions are typified by tall cells with deeply eosinophilic cytoplasm, elongated nuclei bearing delicate chromatin, ectopic crypt foci, deep clefting of the lining mucosa and an overall resemblance to small bowel mucosa.
Broadly, TSAs arise via three mechanisms. They may be BRAF mutated and CpG island methylator phenotype (CIMP)-high: right sided, mediated through a microvesicular hyperplastic polyp or a sessile serrated adenoma, may also have RNF43 mutations and result in microsatellite stable (MSS) colorectal cancers (CRC). The second pathway that is mutually exclusive of the first is mediated through KRAS mutation with CIMP-low TSAs. These are left-sided TSAs, are not associated with another serrated polyp and result in MSS CRC. These TSAs also have RSPO3, RNF43 and p53 mutations together with aberrant nuclear localisation of β-catenin. Third, there is a smaller group of TSAs that are BRAF and KRAS wild type and arise by as yet unknown molecular events. All TSAs show retention of mismatch repair proteins.
Conclusion These are characteristic unusual polyps with a complex molecular landscape.
- traditional serrated adenoma
- serrated polyp
- BRAF
- KRAS
- colorectal cancer
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Recent investigations into the pathogenesis of colorectal carcinoma (CRC) have identified the serrated pathway of colorectal carcinogenesis, which accounts for 15%–35% of CRCs.1–4 There are several potentially different precursor lesions implicated in the serrated pathway that fit into the general category of ‘serrated adenomas’. The term ‘serrated adenoma’ was introduced by Longacre and Fenoglio-Preiser, and later refined initially by Torlakovic et al, and then by others, to incorporate a spectrum of lesions, namely hyperplastic polyps (HP), sessile serrated adenomas/polyps (SSA; also called sessile serrated lesions in the UK and parts of Europe), and traditional serrated adenomas (TSA).1 5–9
When TSAs were first described by Longacre and Fenoglio-Preiser, they described mixed hyperplastic adenomatous polyps/serrated adenomas.5 10 The authors appreciated that, when compared with HPs, these polyps had certain differences, namely prominent nucleoli, goblet cell immaturity, and absence of a thickened basement membrane.5 As awareness of the characteristic morphology of TSAs grew, they are now considered the most unique and easily identified of the serrated lesions.11
The origin of TSAs is unclear, but some probably arise de novo, while many appear to arise in a precursor polyp, typically microvesicular HP or SSA.1 12–19
The purpose of this review is to provide a brief clinicopathological background of TSAs and present the current state of knowledge regarding their molecular pathogenesis.
Clinical and epidemiological features
TSAs account for <1% of all colorectal polyps in most series, and for 1%–7% of all serrated lesions.5 20–24 TSAs tend to occur in older patients (usually over 50 years of age) and have no significant gender predilection.12 15 17 They are found predominantly in the distal (left) colon, and occur only rarely in the proximal colon.1 Most are less than 10 mm in size.
Morphology
In short, the morphological criteria for diagnosing a TSA include typical cytology (ie, elongated, narrow pencillate nuclei with delicate dispersed chromatin and cytoplasmic hypeeosinophilia), ectopic crypt foci (ECF), and typical slit-like clefted serration.6 At least two of these three features are required (with at least one of these features being present in 50% of the polyp) to render a diagnosis of TSA with the slit-like serration being the most consistent histological feature.12 A reproducibility study identified this lesion as the one with the best kappa statistic of the various serrated lesions.25
Architecture
TSAs have an overall protuberant exophytic configuration, with a complex villous growth pattern. Mucosal protrusions, in the form of a ‘tennis-racquet-like’ enlargement of the tip of a protrusion, have been described as a feature unique to TSA.1
Some TSAs demonstrate a flat growth pattern (‘flat’ TSA),1 with the majority of these polyps being elevated less than twice the height of the normal mucosa and lacking prominent villiform projections.12 They are typically found in the proximal colon,12 and Bettington and colleagues demonstrated that flat TSAs can be reliably distinguished from SSAs.12
Cytological features
The lesional cells have centrally placed, elongated, penicillate nuclei (typically not hyperchromatic) (figure 1A).3 5 These lesions are usually not mitotically active, as determined by mitotic count or Ki67 immunoreactivity.1 11 26
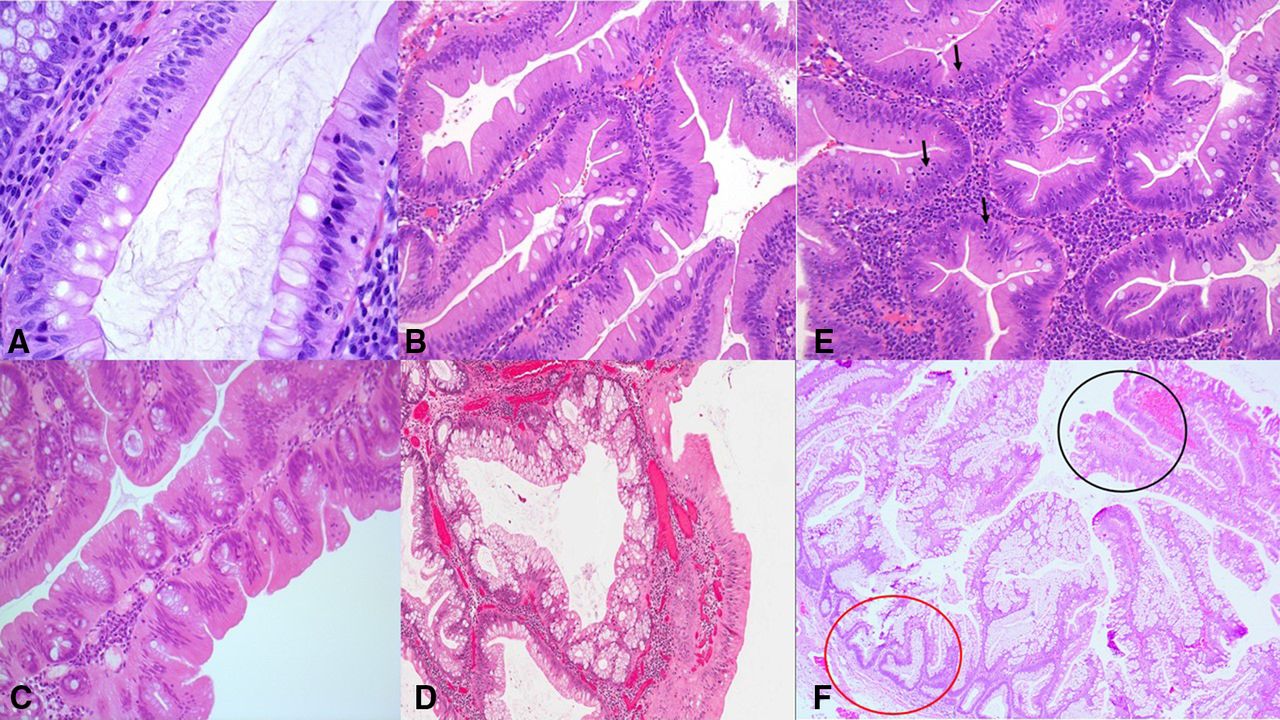
The lesional cells of a traditional serrated adenoma (TSA) have centrally placed, elongated, penicillate nuclei, with dispersed chromatin (A). Narrow slits in the epithelium (‘slit-like serrations’) similar to normal small intestinal mucosa (abundant eosinophilic/oncocytic cytoplasm) are possibly the most specific feature of a TSA (B). Ectopic crypt foci (ECF) refer to the abnormal development of crypts with loss of orientation towards the muscularis mucosae (C). ‘Mucin-rich TSAs’ have 50% or more goblet/mucin-rich cells, with a goblet cell:eosinophilic, absorptive cell ratio of at least 1:1 (D). TSAs have a relatively increased number of intraepithelial lymphocytes (arrows) (E). Many TSAs (black circle) contain adjacent areas of hyperplastic polyp or sessile serrated adenoma (SSA) (red circle), with these latter components being thought to represent a precursor polyp in this setting (F).
The authors have appreciated that the lesional cells in TSAs more or less resemble the normal mucosa of the duodenum, having a brush-border, indented, flat-topped luminal slit-like serrations, pencillate nuclei and eosinophilic cytoplasm.27
Serrations
Slit-like serrations refer to narrow slits in the deeply eosinophilic epithelium similar to normal small intestinal mucosa.4 It has been suggested that these slit-like serrations (figure 1B) are possibly the most specific feature when faced with the challenge of differentiating a TSA from a morphologically similar tubulovillous adenoma with prominent serration.28
Ectopic crypt foci
ECFs refer to the abnormal development of crypts with loss of orientation towards the muscularis mucosae.29 These ectopic crypts are not present in all areas of all TSAs but should be present at least focally, and, in fact, it is thought that they are probably the best defining feature of TSAs (figure 1C), although most TSAs will also have the atypical eosinophilic cell.1 3 13
Once ECFs and slit-like serrations become identifiable in a small (<10 mm) polyp, the diagnosis of a TSA should be considered, even if villous change is not apparent.30
Other features
Goblet cells are not infrequent in TSAs and their amount varies greatly from case to case, as well as from area to area in the same polyp. Some TSAs are composed mainly of mucin-filled goblet cells (‘mucin-rich TSA’),3 31 32 and these mucin-rich TSAs have been arbitrarily defined by the presence of 50% or more goblet/mucin-rich cells with a goblet cell:eosinophilic, absorptive cell ratio of at least 1:1 (31) (figure 1D).
TSAs have been shown to have a relatively increased number of intraepithelial lymphocytes (figure 1E), as compared with classical adenomas, but much less than that of SSAs harbouring conventional adenomatous dysplasia.27 Furthermore, the mucin-rich TSAs have been shown to have more intraepithelial lymphocytes than classic TSAs.31
TSAs that occur in the right colon tend to occur more frequently in females, tend to be more of the mucin-rich variety and have greater intraepithelial lymphocytes than their left-sided counterparts.31 Furthermore, right-sided TSAs are usually BRAF mutated and have methylation of CpG islands (CpG island methylator phenotype (CIMP) positive)—see molecular discussion below.
Precursor polyp
Many TSAs contain adjacent areas of HP (characterised by glands with saw-toothed, serrated luminal profiles and straight or V-shaped basal or lower one-third gland profiles) or SSA (glands which are also saw-toothed but with very characteristic basal architecture: instead of being straight the basal aspects of glands are dilated, fan out horizontally and give rise to boot-shaped or L-shaped glandular profiles), thought to be a precursor polyp in this setting12 17 27 33 (figure 1F). In a study exploring the association of precursor polyps with TSA, 28 were HPs (36%) and 18 were SSAs (23%).33 Thus, up to one-third of TSAs may contain histological evidence of an HP or SSA suggesting a relationship among all three types of serrated polyps. Evidence of one of these precursor components is characterised by a discrete area of the lesion with clear morphological distinction from the TSA component, either at the edge of, or underlying, the TSA.15
Dysplasia
Although some authors interpret the prototypical lesional cell of TSA as being inherently dysplastic, others maintain that this cell type itself is generally non-proliferative and therefore may, in fact, represent a metaplastic or a senescent cell.1 3 4 10–12 18 19 26 34–42
The presence of an abrupt transition from typical TSA to discrete areas of cytological atypia or dysplasia of a more conventional type can be seen with varying frequency. The conventional dysplasia is often found towards the base of the TSA,1 3 15 43 44 and it has been proposed that these lesions be referred to as ‘TSAs with conventional cytological dysplasia’3 or ‘advanced TSAs’.12 Bettington and colleagues showed that advanced TSAs tend to be larger than ordinary TSAs.12
Conventional dysplasia
Cytological features of conventional adenomatous dysplasia include increased nuclear size, nuclear crowding, hyperchromasia, complete loss of polarity, and pseudostratification with nuclei extending into the upper half of the neoplastic cell, in addition to frequent and atypical mitoses.35 43 44 Architectural features of conventional adenomatous dysplasia include crowding of glands, cribriform glands, and intraluminal necrosis.35
Serrated dysplasia
Serrated dysplasia, as defined by the WHO, is characterised by cuboidal cells with eosinophilic cytoplasm, vesicular nuclei and prominent nucleoli.35
For now, assigning a grade of dysplasia or dividing dysplasia into serrated versus conventional types has no clinical utility and the practising gastroenterologist should not treat a TSA with low-grade dysplasia any differently to a TSA without overt dysplasia and one-off surveillance colonoscopy at 3 years should be performed.12 45
Biomarker expression
TSAs do not express a wide range of routine biomarkers and there are no specific markers available yet. With regard to some of the newer biomarkers such as annexin A10, Hairy and Enhancer of Split 1 (Hes-1) and SPARC-related Modular Calcium-binding 1 protein (SMOC1) more studies are required to confirm their applicability in TSAs.
Ki-67 and CK20
In TSAs, Ki-67 expression is limited to the ECF and basal aspects of the crypts. The opposite staining pattern is found with CK20 where expression is almost exclusively limited to the superficial lining of the surface of the polyp, without extension into the budding crypts.1 12
Annexin A10 and Hes-1
Recent studies have shown that expression of annexin A10 is a reliable marker of SSAs and the serrated pathway of CRC,16 17 46–51 and loss of Hes-1 expression can reliably differentiate SSAs from HPs.52 Nourbakhsh and Minoo sought to interrogate the concept that at least some TSAs may arise in association with precursor HP or SSA lesions, particularly those that develop in the right colon, by applying these two stains to a series of polyps from the right side of the colon with mixed features of TSA and SSA (as defined by Rex et al).7 16 The TSA components of these ‘hybrid or mixed polyps’ showed a staining pattern similar to that of SSAs (annexin A10 overexpression and Hes-1 loss), which the authors suggest supports the theory that SSAs are precursor lesions for at least some TSAs.16
SPARC-related Modular Calcium-binding 1 protein
Aoki et al showed that SMOC1 is expressed immunohistochemically (cytoplasmic staining) in normal colonic epithelium and in SSAs, but its expression is decreased in TSAs.53 Thus, they suggested that immunohistochemical staining of SMOC1 is highly discriminative between TSAs and SSAs, and could be used as an ancillary tool in challenging polyps. They also demonstrated that SMOC1 is frequently methylated in TSAs and rarely methylated in SSAs.53
p53/p16/β-catenin
Bettington and colleagues showed that advanced TSAs (defined as those with overt dysplasia or carcinoma) displayed strong p53 staining in just over half of cases,12 which is an adequate surrogate for TP53 gene mutation.
They demonstrated nuclear β-catenin staining in 40% of cases, indicative of Wnt pathway activation as an important step in malignant progression.12
The authors also showed loss of p16 staining in the advanced areas of just over half of BRAF-mutant TSAs, and of one-tenth of KRAS-mutant TSAs or BRAF/KRAS-wild-type TSAs.12 This loss is attributed to methylation-induced silencing of the CDKN2A gene which appears to be an important step in the development of adenocarcinoma in these polyps.12
Mismatch repair proteins
Mismatch repair (MMR) enzyme function is retained in effectively all TSAs, even when they develop high-grade dysplasia or invasive adenocarcinoma, implying a microsatellite stable (MSS) phenotype.12 44
MMR deficiency has, however, been demonstrated in TSAs from proven MMR gene mutation carriers, with loss of immunohistochemical expression in keeping with the underlying germline mutation.54 High-level microsatellite instability (MSI-H) results were shown to be concordant with MMR protein loss results in these polyps.54 None of the cases tested (all of which were MLH1-deficient polyps) demonstrated MLH1 methylation or somatic BRAF c.1799T>A (p.V600E) mutation.54
Very rarely, sporadic TSAs do display MLH1 hypermethylation.12 27 Bettington et al showed in their series of 200 TSAs that MLH1 promoter methylation was present in 7% of the BRAF-mutant TSAs (eight ordinary/classical and one advanced), but in none of the KRAS-mutant, or BRAF/KRAS-wild-type TSAs.12 Only the advanced TSA with MLH1 methylation showed concordant loss of MLH1 expression by immunohistochemistry.12
Molecular changes
As previously stated, it is now known that 15%–35% of CRCs arise as a consequence of the serrated neoplasia pathway.2–4 Mitogen-activated protein kinase (MAPK) pathway activation, a critical early event as a consequence of either activating BRAF or KRAS mutation,55 and the CIMP, causing methylation of CpG islands in the promoter regions of several genes resulting in gene silencing, are well-established molecular drivers of this pathway.55–57
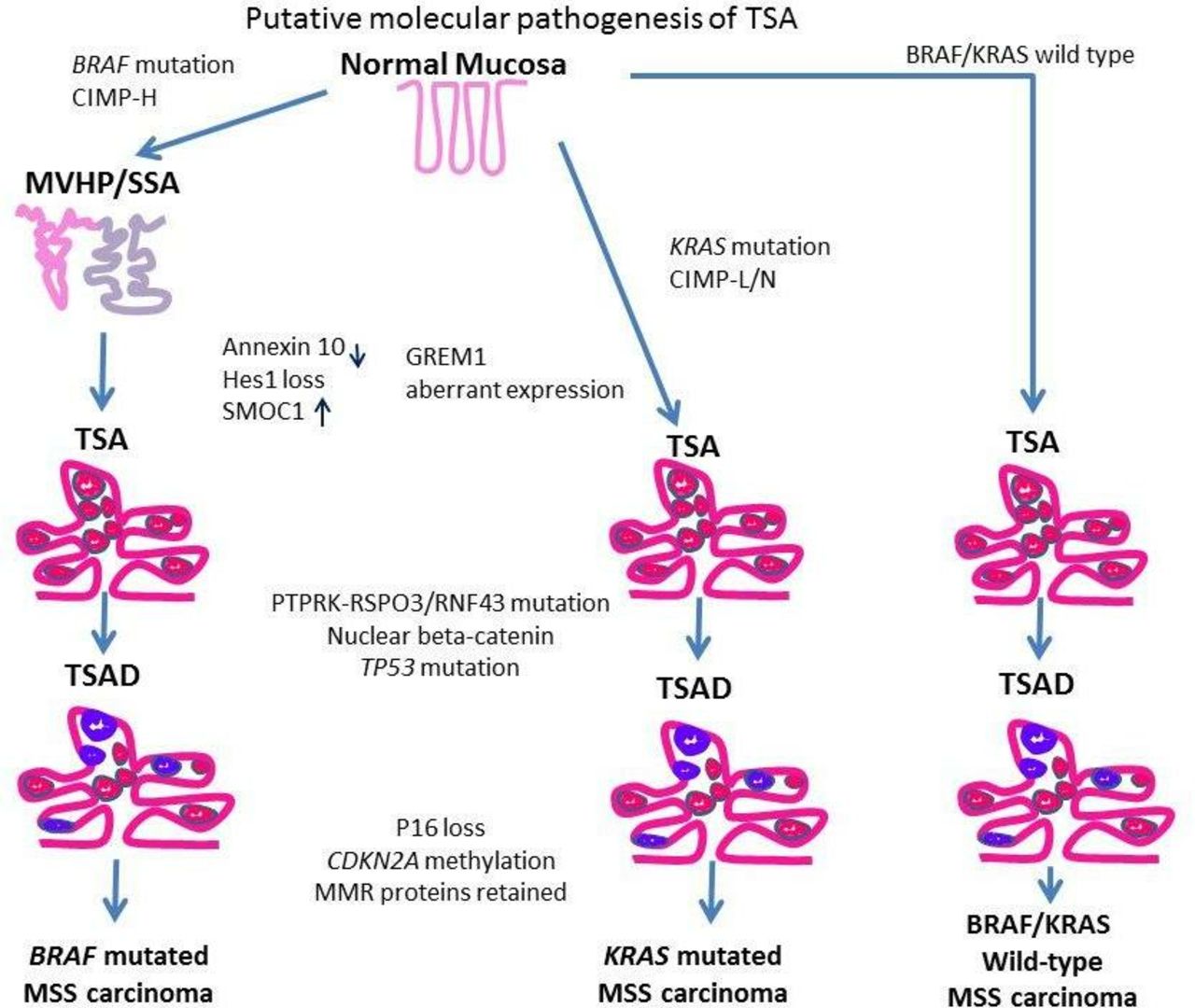
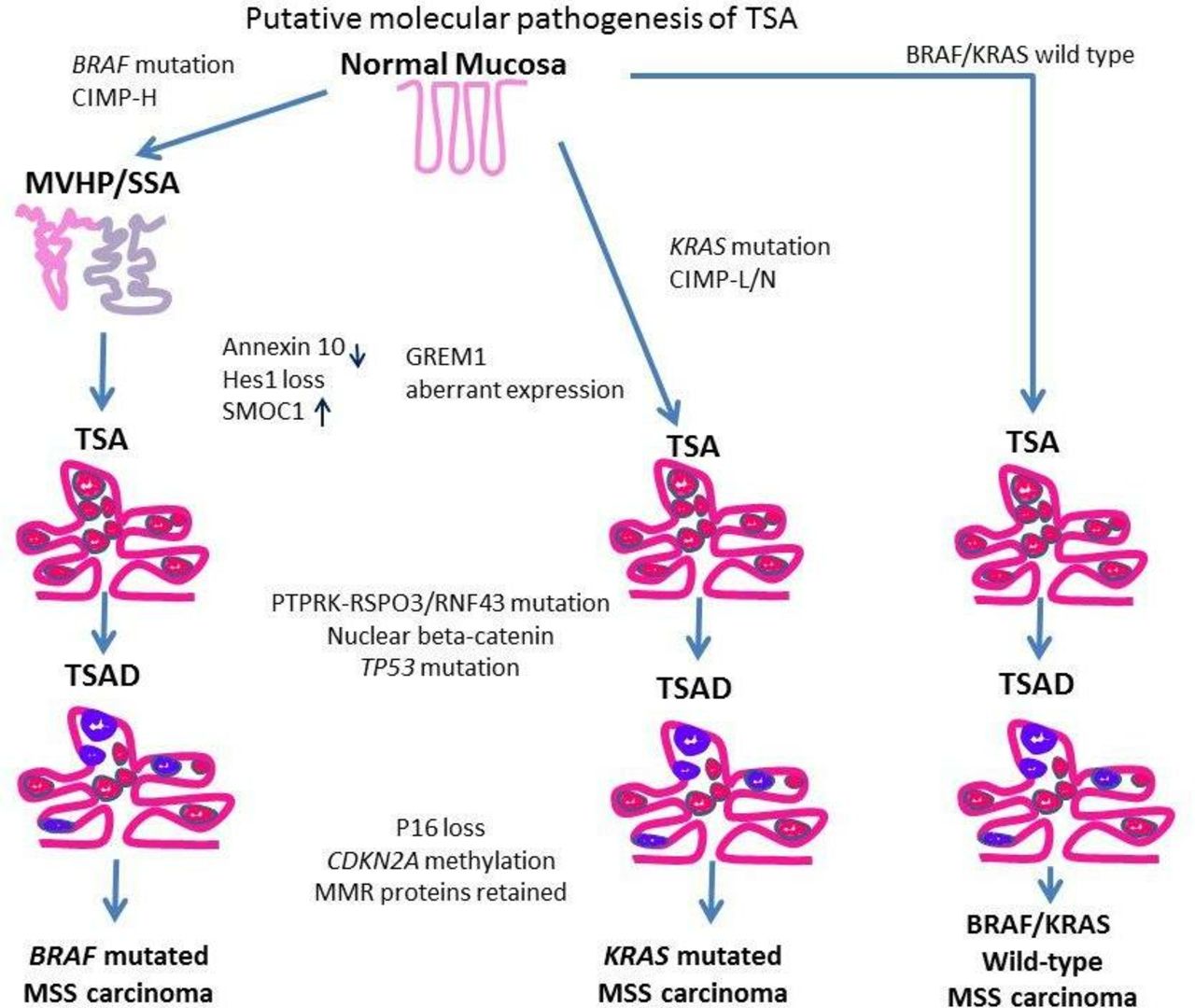
TSAs are thought to arise via three molecular pathways.
The first mechanism is via BRAF mutation and CpG island methylation (CIMP) resulting in the CIMP-high (CIMP-H) phenotype. These TSAs tend to be right sided, maybe associated with a precursor microvesicular HP, or an SSA. Furthermore, they may also have RNF43 mutations and result in MSS colorectal cancers (CRC). The second pathway that is mutually exclusive of the first is mediated through KRAS mutations and these are CIMP-low (CIMP-L) TSAs. These TSAs are usually located in the left colon, are not associated with another precursor serrated polyp but also result in MSS CRC like the first pathway described above. These TSAs may also show RSPO3, RNF43 and p53 mutations together with aberrant, nuclear localisation of β-catenin.
The third pathway resulting in a smaller group of TSAs is both BRAF and KRAS wild type and arise by, as yet, unknown molecular events.
Although TSAs are genetically a heterogeneous group they are recognised as a precursor lesion for MSS or low-level MSI CRC and, only rarely lead to sporadic MSI-H colon cancers.4 19 This even applies to those TSAs harbouring a BRAF mutation that are mostly located in the right colon.12 15 18
MAPK pathway
Studies have reported either BRAF or KRAS mutations in the vast majority (>80%) of TSAs,27 and BRAF (22%–42% of TSAs) and KRAS (48%–67% of TSAs) mutations are mutually exclusive.14 15 17 27 37 43 44
KRAS mutations occur predominantly at codon 12 and less frequently at codon 13, and the most common mutations are G12D, G12V and G13D occurring in 0%–28% of TSAs.20 57 58 With regard to BRAF, the most frequent mutation is V600E which occurs in 60%–76% of TSA.20 57–60
It has been shown that, although BRAF-mutated and KRAS-mutated TSAs are molecularly different, these lesions show the same morphology.27 44 61 However, BRAF-mutant TSAs are more often located in the proximal colon and are thought to have more frequent origin in an associated precursor polyp than KRAS-mutant TSAs.12 On the other hand, KRAS-mutant TSAs are almost exclusively located in the distal colon, especially the rectum.12 Furthermore, most mucin-rich TSAs, which are typically right sided, also harbour a BRAF mutation.32 A small proportion of TSAs are BRAF/KRAS wild type, and these cases segregate best with the KRAS-mutated group.12
CpG island methylation
CIMP-H status has been shown to correlate strongly with both proximal colonic location and BRAF mutation.12
It has been shown that BRAF-mutant TSAs are more often CIMP-H than either KRAS-mutant, or BRAF/KRAS-wild-type TSAs, while the majority of KRAS-mutant, or BRAF/KRAS-wild-type TSAs are either CIMP-L or negative for CIMP.12 44 Furthermore, TSAs found in the proximal colon are more likely to be CIMP-H, compared with those in the distal colon.12 44 Also, in the distal colon, BRAF-mutant TSAs are more likely to be CIMP-H than KRAS-mutant TSAs.12 44 62
Wnt pathway
Wnt pathway activation, resulting from Protein Tyrosine Phosphatase, Receptor-type K-R-Spondin 3 (PTPRK-RSPO3) gene fusions or Ring Finger Protein 43 (RNF43) gene mutations, occurs in the majority of TSAs.62RSPO fusions and RNF43 mutations are characteristic of TSA and are rare or absent in other types of colorectal polyps.63 64
RSPO fusions
The PTPRK-RSPO3 fusions in TSAs are mutually exclusive with other Wnt pathway gene alterations, and are thought to be responsible for Wnt pathway activation,63 potentiating ligand-dependent Wnt signalling.65 Sekine and colleagues showed that RSPO fusions were rarely observed in TSAs located in the proximal colon.66 They showed that TSAs with RSPO fusions tended to be larger, and to have ECF and a high-grade dysplastic component; while slit-like serrations were less prominent, and associations with precursor polyps being rare in these RSPO fusion-positive TSAs.65 It has been shown that TSAs with RSPO fusions are less likely to have BRAF mutations, and more frequently have KRAS mutations.63 66
RNF43 mutations
RNF43 mutations have been detected in over 25% of TSAs.63 64
Most of the RNF43 mutations that have been detected in the TSAs were homozygous, indicating the biallelic inactivation of these tumour suppressor genes.36
RNF43 mutations have also been detected in a small number of TSA-precursor polyps.36 63 64 67 It has been demonstrated that RNF43 mutations in the precursor polyps are heterozygous, in contrast to the homozygous mutations found in the established TSA components,36 suggesting that biallelic inactivation of RNF43 occurs during the progression to TSAs in these lesions.36
Germline mutations of RNF43 have been identified in a minority of patients satisfying the WHO criteria of serrated polyposis syndrome in two studies,67–69 leading Yan et al to suggest that routine germline testing for RNF43 mutation should be performed in serrated polyposis syndrome families.67
TSAs with a precursor component
Molecular studies previously done in both components (ie, on the TSA component and on the HP/SSA component) of TSAs with a histologically identifiable precursor component (HP/SSA) confirmed identical mutations of KRAS or BRAF in both components, indicating that MAPK pathway gene mutations are the earliest molecular abnormality occurring in the serrated pathway of tumourigenesis.14 15 36
BRAF mutations were more commonly seen in TSAs with an identifiable precursor component located in the right colon, supporting the hypothesis that HPs/SSAs are indeed precursors for TSAs.14 Thus, it has been suggested that in a BRAF-mutated SSA, methylation of MLH1 takes the oncogenic pathway to MSI-H CRCs, with the SSA progressing to an intermediate stage of SSA with cytological dysplasia before full-blown malignancy.16 However, it is believed that if Wnt pathway becomes activated (eg, by RNF43 mutation) before methylation extends to the MLH1 locus, then the SSA will transform into a TSA and then the lesion will progress through the MSS pathway and almost never goes back to being MSI-H.16 63 64 67 70
Miscellaneous genes
A variety of other changes at the molecular level have also been identified in a subset of TSAs. While further studies are required to confirm their roles, these include:
p16 hypermethylation
p16 hypermethylation has been shown to be characteristic of TSAs.27
GNAS mutation
GNAS mutation has been identified in a small fraction of TSAs (<10%), mainly with concomitant BRAF mutation,17 63 71 but GNAS mutation status does not correlate with advanced histology (ie, TSAs with high-grade dysplasia and/or invasive carcinoma).17 71 72 Liu and colleagues concluded in their study that no histological features separate GNAS-mutant TSAs from GNAS-wild-type TSAs.71
Significance
There is evidence that some of these serrated lesions lead to certain subtypes of CRC, which account for interval carcinomas found during endoscopic surveillance programmes and which are biologically different from the classical Vogelstein model for CRC.27 74 A 3-year colonoscopy surveillance interval is currently recommended by many guidelines for usual TSA.75–79
A carcinoma arising in TSA can be the typical colorectal adenocarcinoma not otherwise specified, but can also be a mucinous or serrated adenocarcinoma.39 They are often CIMP-H, but are essentially never MSI.
Conclusion
TSA has a characteristic constellation of morphological features that make microscopic recognition of classic examples relatively simple. Histological variants have been described and maintain the basic histological tenets seen in typical TSA. The relative simplicity of morphology is counterbalanced by the molecular complexity that TSAs manifest (figure 2). As we have highlighted, despite the fundamental dichotomy of BRAF versus KRAS-mutated TSA, the morphological appearances remain the same. At this juncture, there are no compelling reasons to believe that the different molecular pathways confer varying propensities for the development of carcinoma.
{kind=link}
{kind=link}
Schematic representation of the putative molecular pathogenesis of TSA showing three pathways. Purple areas represent dysplastic foci. CIMP-H, CpG island methylator phenotype-high; MMR, mismatch repair; MSS, microsatellite stable; SSA, sessile serrated adenoma; TSA, traditional serrated adenoma.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
Footnotes
Contributors The three authors (AJMC, SS, RC) are authors based on the following four criteria applying to all three authors: substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work: AJMC, SS, RC; drafting the work or revising it critically for important intellectual content: AJMC, SS, RC; final approval of the version to be published: AJMC, SS, RC; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: AJMC, SS, RC. All individuals listed as coauthors of the manuscript (AJMC, SS, RC) qualify for every one of the four criteria listed above.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement No additional data are available.