Article Text

Abstract
Background Alcohol is classified as a Group 1 carcinogen by the International Agency for Research on Cancer because it induces hepatocellular carcinoma (among other cancers) in humans. An excessive alcohol intake may result in fatty liver, acute/chronic hepatitis, and cirrhosis and eventually lead to hepatocellular carcinoma. It has been reported that alcohol abuse increases the relative risk of hepatocellular carcinoma by 3- to 10-fold.
Aim and Methods To clarify the known mechanisms of alcohol-related carcinogenesis, we searched Pubmed using the terms alcohol and immune mechanism, alcohol and cancer, and immune mechanism and cancer and summarized the articles as a qualitative review.
Results From a clinical perspective, it is well known that alcohol interacts with other factors, such as smoking, viral hepatitis, and diabetes, leading to an increased risk of hepatocellular carcinoma. There are several possible mechanisms through which alcohol may induce liver carcinogenicity, including the mutagenic effects of acetaldehyde and the production of ROS due to the excessive hepatic deposition of iron. Furthermore, it has been reported that alcohol accelerates hepatitis C virus-induced liver tumorigenesis through TLR4 signaling. Despite intense investigations to elucidate the mechanisms, they remain poorly understood.
Conclusion This review summarizes the recent findings of clinical and pathological studies that have investigated the carcinogenic effects of alcohol in the liver.
- hepatocellular carcinoma
- alcohol
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
Worldwide, liver cancer is the second highest cause of cancer-related death in men and the sixth highest cause of cancer-related death in women. Liver cancer is more common in low-income and middle-income countries than in developed countries.1 According to the National Cancer Institute, approximately 40 700 cases of liver cancer are expected to be newly diagnosed, and approximately 29 000 patients will die from liver cancer in the USA in 2017. Besides, the incidence of liver cancer is increasing by approximately 3%–4% per year.2 This means that liver cancer is a major public health problem.
Hepatocellular carcinoma (HCC), which accounts for around 70%–90% of cases, is the most common type of primary liver cancer. Alcohol consumption, the level of which is higher in developed countries, especially in the USA and Europe,3 is one of the frequent causes of HCC in developed countries.4 Conversely, chronic hepatitis B virus (HBV) infection is the major risk factor in low-income and middle-income countries. The ratio of alcohol abuse to all aetiologies of HCC varies according to the country and area; alcohol abuse is reported to be responsible for approximately 15%–30% of HCC.4 5 However, the appropriate methods for surveilling patients with alcohol use disorder (AUD) to facilitate the early-stage diagnosis of HCC remain to be determined, and mechanisms through which alcohol consumption is involved in the initiation of HCC remain unclear.
Understanding the clinical features and the mechanisms of alcohol-based HCC is critically important to the prevention and detection of early-stage HCC and for the development of treatments for HCC. This review summarises the recent clinical and pathological studies investigating the carcinogenic effects of alcohol in the liver.
The risks of liver cirrhosis and HCC
According to a WHO report, approximately 280 million individuals, or 4.1% of the population aged >15 years, meet the definition of AUD (alcohol dependence and the harmful use of alcohol). The prevalence is almost the same as the prevalence of hepatitis B, and is four times higher than the prevalence of hepatitis C.3 6 Because of the large population—HCC screening (eg, ultrasonography or the measurement of serum tumour marker levels) for all of such patients would lead to huge medical costs—it is necessary to select individuals with a high risk of HCC. In this respect, the American Association for the Study of Liver Diseases (AASLD) recommends that patients with Child’s classification A/B cirrhosis undergo surveillance for HCC using ultrasonography with or without alpha-fetoprotein measurement, every 6 months, and does not recommend the modification of the surveillance strategy based on the ‘etiology of liver disease’, the strategy of which is almost the same as that recommended by the European Association for the Study of the Liver.7 8 Incidentally, the previous AASLD guidelines for the management of HCC suggested that HCC surveillance is cost-effective if the annual incidence of HCC is ≥1.5% in patients with cirrhosis. Similar to hepatitis C and hepatitis B, the presence of alcoholic liver cirrhosis is considered to be an important risk factor for the development of HCC. It has been reported that approximately 10%–20% of heavy drinkers develop cirrhosis.9 Furthermore, several previous studies that have assessed the annual incidence of HCC in patients with alcohol-induced liver cirrhosis have revealed the rate to be 1.9%–2.6%.10 11 Thus, it might be appropriate to perform HCC surveillance for patients with alcoholic liver cirrhosis. However, even when guideline-based surveillance was performed, almost 20%–30% of HCC in patients with cirrhosis were diagnosed at a non-early stage.12 It is therefore necessary to determine the traits of patients with alcoholic liver cirrhosis that increase their risk of HCC and the traits of patients with AUD who show the highest risk of HCC.
The amount of ethanol as a risk factor
An excessive alcohol intake has been shown to be a risk factor for liver cirrhosis and HCC. It is considered that there is a linear dose–response relationship between alcohol consumption and the risk of cirrhosis and HCC.13 An alcohol intake of 30–50 g/day increases the OR for liver cirrhosis,14–16 while an alcohol intake of >60–100 g/day increases the OR for HCC.17–20 In terms of the total amount of alcohol intake during a patient’s lifetime, an alcohol consumption of >600 000 mL significantly increased the risk of HCC development (OR=4.52, 95% CI 2.39 to 8.55).21 22 Thus, the amount of ethanol consumption might provide an indication of the risk of liver cirrhosis and HCC.
Gender differences as a risk factor
There might be a gender difference in the volume of alcohol intake that increases the risk of alcohol-induced liver damage and the development of HCC. It has been reported that the risk of developing cirrhosis becomes substantial with the consumption of 60–80 g/day of alcohol for 10 years in men and 20 g/day for 10 years in women.14 23 In addition, women showed a more rapid progression (20 years) to cirrhosis than men (35 years).24 Among individuals who consume more than 80 g/day of alcohol, the risk of HCC development in women has been shown to be almost fivefold higher than that in men.18 However, the overall prevalence of HCC in women is small compared with that in men.
Various mechanisms have been suggested to underlie the higher sensitivity of women to alcohol.25 After the oral administration of alcohol, women show less first-pass metabolism of alcohol, which is defined as the difference in the amount of orally administered ethanol and the quantities in the systemic blood, due to their lower gastric alcohol dehydrogenase (ADH) activity, which results in a higher serum concentration of alcohol.26 Thus, even when the same amount of ethanol is consumed, the female liver may be exposed to more ethanol. Furthermore, oestrogen (a female sex hormone) may play an important role in alcohol-induced liver injury. It has been shown that oestrogen increases the sensitivity of Kupffer cells to lipopolysaccharide (LPS), which results in more severe liver injury.27 28 In fact, many previous studies have reported that more severe inflammatory responses in the liver and fat tissue, which were associated with toll-like receptor (TLR4) signalling, were seen in female patients.29–35
Conversely, several lines of evidence indicate that oestrogen and its downstream signalling protect against HCC development.36–38 In N-nitrosodiethylamine (DEN)-induced HCC mouse model, it has been shown that ablation of interleukin-6 (IL-6) abolished the gender differences in HCC development, and oestrogen attenuates serum IL-6 levels and IL-6 mRNA levels of Kupffer cell. These results suggest that oestrogen reduces the risk of HCC by the inhibition of IL-6 production from Kupffer cells.39 Taken together, the gender disparity in sensitivity to alcohol and HCC development cannot be sufficiently explained by simple mechanism. Although the prevalence of alcoholic liver disease (ALD) in female patients is low,3 they may require more intense surveillance.
Given that the importance of LPS–Kupffer cell interaction has also been reported in a non-alcoholic steatohepatitis (NASH) model,40 41 young females could be severely affected with NASH via oestrogen-related LPS–Kupffer cell interaction worsening. However, several studies have demonstrated that oestrogen protects against liver inflammation and fibrosis in a NASH model, and oestrogen deficiency has been reported to worsen steatohepatitis and liver fibrosis in human.42 43 As oestrogen has multiple functions and the expression profile of the mechanisms might differ in patients’ status, it is difficult to explain the effect simply.
The coexistence of hepatitis virus as a risk factor
In patients with ALD, the coexistence of hepatitis virus has been shown to accelerate the disease course.44 It has been reported that the prevalence of hepatitis C virus (HCV) infection in alcoholic patients is 16.32%,45 which is much higher than that in the general population (1.5%–2.0%).46 47 In patients with a high alcohol intake (>60 g/day to 125 g/day), the coexistence of HCV has been shown to increase the risk of alcohol-associated liver cirrhosis.17 18 24 48–50 Furthermore, heavy alcohol consumption has also been shown to increase the risk of developing HCC.18 44 Patients with coexisting HBV (defined by (hepatitis B s antigen) HBsAg-positivity) are at increased risk of developing fibrosis48 and HCC.51 52 In addition, self-resolved HBV infection (defined as HBsAg-negative, HCVAb-negative and HBcAb-positive) can be a risk factor for developing HCC in patients with alcoholic cirrhosis.53 Although the mechanisms by which the synergistic effect between alcohol and hepatitis virus increases the risk of liver fibrosis and HCC have been the subject of extensive research, they have not been completely elucidated.54–59 Collectively, it might be better to perform surveillance of patients with ALD who show coexisting HBV or HCV.
Diabetes and obesity as risk factors
It has been well recognised that diabetes is a risk factor for HCC. Several meta-analyses have shown that diabetes is significantly associated with HCC (relative risk: 1.87–2.32).60–64 As for the association between obesity and HCC, Larsson and Wolk61 reported that the relative risk of liver cancer among obese (defined as a body mass index of >30) individuals was 1.89 (95% CI 1.51 to 2.36).61 Case–control studies have reported a synergistic interaction between heavy alcohol consumption and diabetes that affects the risk of HCC (OR: 4.2–9.9).44 65 Correspondingly, alcohol use and obesity showed a synergistic interaction with the risk of developing HCC (HR: 3.82, 95% CI 1.94 to 7.52).66 Although the mechanisms of the synergistic effect between ALD and diabetes are unknown, several mechanisms have been suggested to underlie the development of diabetes and/or obesity-induced HCC in patients with non-alcoholic steatohepatitis.67 In terms of cost-effective surveillance, a recent cohort study investigated the potential predictors of HCC in 3544 patients with diabetes without viral hepatitis and suggested that a DM-HCC risk score included age >65 years, low triglyceride levels and high gamma-glutamyl transferase levels.68 In this study, although heavy drinking was found to be a significant predictor in a univariate analysis, it did not remain significant in a multivariate analysis.
In addition to the above-mentioned risk factors, others have been suggested, including genetic polymorphisms,69 race and ethnicity.70 Thus far, unfortunately, there are no definitive hallmarks to narrow down the target patients. Further studies are required to establish clinically beneficial prediction models.
Potential mechanisms of alcohol-induced HCC
The mechanisms underlying the induction of carcinogenesis by alcohol have not been fully elucidated. Because there are no animal models of alcohol-induced HCC alone, a chemical-induced (DEN) HCC model is mainly used to study the effects of alcohol intake on hepatocarcinogenesis.32 71 72 However, the priming effect of hepatocarcinogenesis and the progressive effects have been well investigated.
Alcohol absorption and metabolism
Once ethanol is consumed, almost all of it is absorbed by the small intestine and metabolised by the liver.73 Ethanol is metabolised to acetaldehyde by ADH in the cytoplasm of hepatocytes, acetaldehyde subsequently enters the mitochondria, and is then oxidised to acetate by mitochondrial aldehyde dehydrogenase (ALDH). When a large amount of ethanol is consumed, cytochrome P450 2E1 (CYP2E1), which mainly exists in the endoplasmic reticulum, and catalase of peroxisomes also contribute to the metabolism of ethanol. The CYP2E1-dependent pathway can catalyse ethanol into acetaldehyde while producing reactive oxygen species (ROS), such as hydroxyethyl, superoxide anion and hydroxyl radicals74 (figure 1). In humans, there are at least seven different isozymes of ADH and four isozymes of ALDH. Several previous studies showed ALDH2 polymorphisms to be significantly associated with the development of HCC75; thus, it is possible that the metabolism of alcohol is involved in the mechanisms of HCC development.
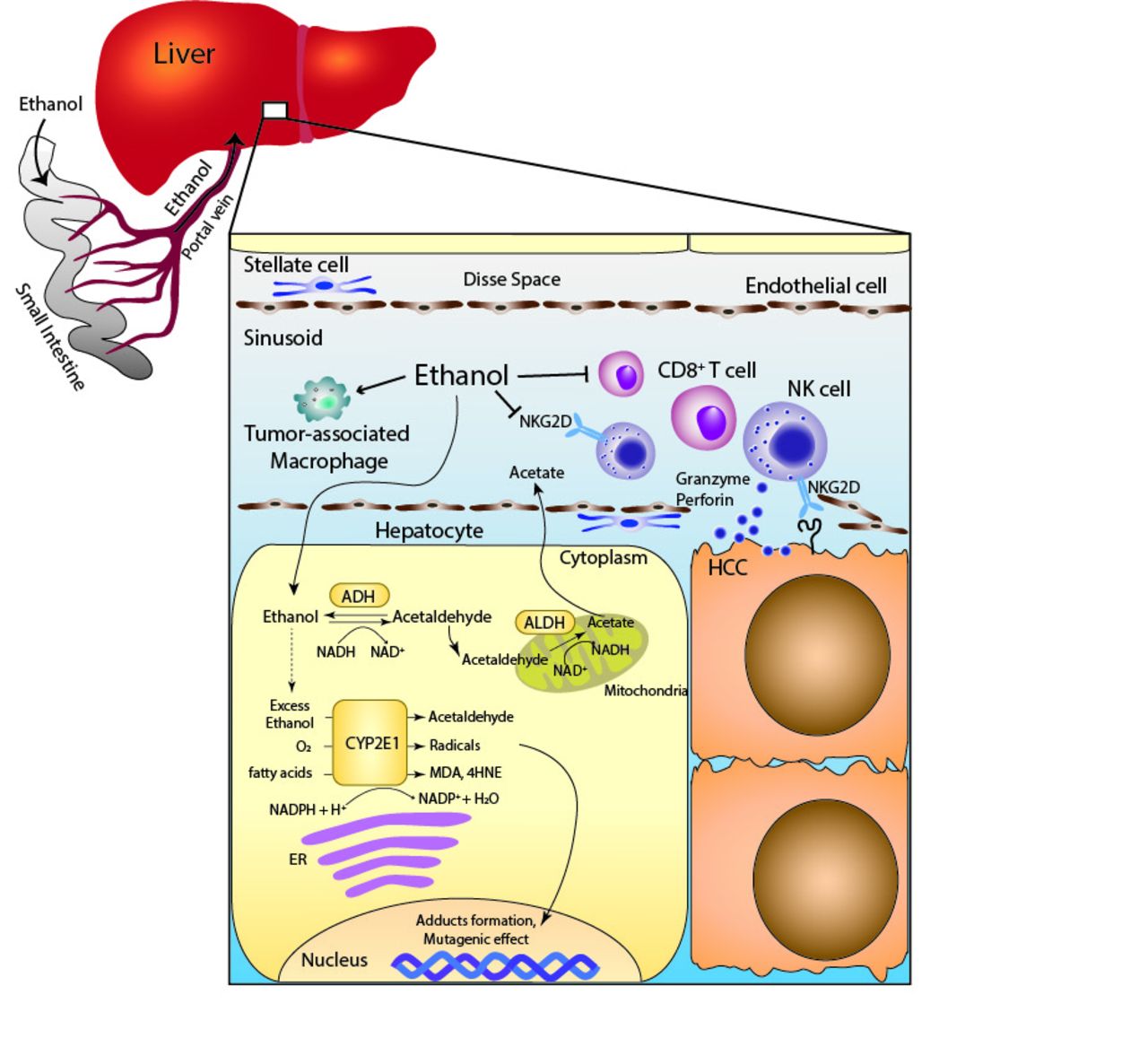
{kind=link}
Scheme of the immune system in HCC surveillance and the metabolic effects of alcohol exposure on hepatocyte. The metabolism of ethanol through the CYP2E1-dependent pathway produces acetaldehyde, radicals and lipid peroxidation products, such as MDA and 4HNE. Alcohol consumption reduces the number of CD8+ T cells and NK cells, and reduces the NKG2D expression on NK cells. 4HNE, 4-hydroxy-2-nonenal; ADH, alcohol dehydrogenase; ALDH, aldehyde dehydrogenase; CYP2E1, cytochrome P450 2E1; ER, endoplasmic reticulum; HCC, hepatocellular carcinoma; MDA, malondialdehyde; NK, natural killer; NKG2D, NK group 2D.
The carcinogenic properties of acetaldehyde, an alcohol metabolite
Acetaldehyde has been shown to be a carcinogen in animal studies. With regard to direct DNA mutagenic mechanisms, it has been reported that acetaldehyde increases the point mutation frequency in the hypoxanthine phosphoribosyltransferase (HPRT) gene in lymphocytes,76 and induces sister chromatid exchanges.77 Meanwhile the formation of adducts with DNA, for example, N2-ethyl-deoxyguanosine (N2-Et-dG),78 has been found in patients with ALD. Another DNA adduct, N2-propano-2’-deoxyguanosine (N2-Et-dGTP), has been reported to cause the alternation of DNA integrity.79 Furthermore, the formation of protein adducts plays an important role in carcinogenesis. Acetaldehyde interacts with certain amino acids in proteins, for example, the formation of adducts with O6-methylguanine methyltransferase causes DNA repair system dysfunction.80 With regard to liver fibrosis, acetaldehyde produced in the hepatocytes can enter the hepatic stellate cell (HSC) and induces the expression of type I collagen genes in vitro.81 Acetaldehyde adducts with protein have been shown in HSC.82 Taken together, the direct DNA mutagenic effect of acetaldehyde and the indirect carcinogenic effect through the formation of adducts may modulate hepatocarcinogenesis and liver fibrosis.
Alcohol and oxidative stress
As described above, the metabolism of ethanol through the CYP2E1-dependent pathway produces ROS. Subsequently, the increase of ROS results in the generation of lipid peroxidation products, such as malondialdehyde (MDA) and 4-hydroxy-2-nonenal (4HNE).83 A significant increase in CYP2E1 could be induced for 1 week in patients who consumed more than 40 g of ethanol per day and was further elevated after 4 weeks.84 In an animal model, the induction of CYP2E1 by ethanol causes the production of hydroxyethyl radicals and lipid peroxidation.85 Furthermore, in humans, it has been shown that ethanol-mediated CYP2E1 induction leads to the increased levels of ROS, lipid peroxidation, MDA and 4HNE.86 4HNE, one of the lipid peroxidation products, can cause a mutation at codon 249 of the p53 gene87; this mutation is commonly found in HCC (20%–30%). In addition to the mutagenic effects on DNA, ROS play an important role as mediators of tumour angiogenesis and metastasis. It has been shown that alcohol-induced ROS production results in the activation of NF-kB signalling; conversely the expression levels of VEGF and MCP-1, the tumour growth, angiogenesis and metastasis were suppressed by the inhibition of alcohol-mediated ROS production and NF-kB signalling.88 Apart from above-mentioned mechanisms of CYP2E1-mediated ROS generation, iron overload and tumour necrosis factor-alpha from inflammatory cells also influence ROS production. Chronic ethanol consumption increases intestinal iron absorption and hepatic iron storage. Iron overload has been shown to cause DNA strand breaks and p53 mutation, which could cause hepatocarcinogenesis.89 90
Alcohol and the methylation of DNA and protein
Aberrant DNA methylation and protein methylation may play important roles in the development of various cancers, including HCC. In most cases, DNA methylation is associated with a decreased gene expression because of interference with the interaction of transcription factor and CpG islands of promoter lesions (usually unmethylated).91 It has been shown that alcohol inhibits the synthesis of S-adenosyl-L-methionine (SAMe), which is a universal methyl group donor. The generation of SAMe is induced by the enzyme methionine adenosyltransferase (MAT). MAT is encoded by two different genes: MAT1A and MAT2A. It has been shown that MAT1A knockout mice develop SAMe deficiency, fatty liver and HCC.92 Furthermore, in patients with AUD, the hepatic MAT activity and the expression of the MAT1A gene are reported to be decreased.93 SAMe also works as a methyl group donor for protein methylation. A recent study by Jie Zhao et al94 reported that the activity of PRMT1 in the mouse liver, which is a protein arginine methyltransferase, was inhibited by ethanol feeding. They revealed that the Hnf4α-dependent hepatocyte proliferation was regulated by the balance of methylation and demethylation of Hnf4α promoter, which depends on PRMT1 and JMJD6, respectively.94
Taken together, elucidating the mechanisms of the aberrant DNA and protein methylation in HCC may lead to the discovery of novel therapeutic targets, and SAMe might be a potential candidate for preventive medicine targeting HCC.
The tumour microenvironment
Recent studies have highlighted the cross-talk between tumour cells and the surrounding microenvironment; in the liver, this mainly consists of immune cells, HSC, fibroblasts and sinusoidal endothelial cells. Although there is increasing evidence to show that the tumour microenvironment influences the development of HCC,95 the mechanisms by which alcohol consumption contributes to the progression of HCC remain unclear. In this respect, recent studies have reported that ethanol feeding promoted DEN-induced tumourigenesis in the liver, and reduced the number of antitumour CD8+ T cells and increased the number of tumour-associated macrophages and/or M2 macrophages in mouse models.96 97 These studies strongly indicate that the immune systems of alcohol-induced HCC may also be a potential therapeutic target.
The role of the immune system in tumour surveillance
Natural killer (NK) cells, which are characterised by CD56+ and CD3− lymphocytes in humans, seem to play a role in tumour surveillance. NK cells are abundant in the liver, where they can constitute up to 30% of the intrahepatic lymphocyte population.98 The release of granules (ie, perforin and granzymes) and the secretion of cytokines (ie, Fas and TRAIL) by NK cells have a rapid cytotoxic effect. NK cells express several activating receptors, such as the NK group 2D (NKG2D), natural cytotoxicity receptors, CD226 (DNAX accessory molecule-1) and CD16 (Fcγ receptor III), to recognise ligands. There is increasing evidence to suggest that NK cells contribute to the pathogenesis of HCC.99 In clinical studies, patients with HCC with low levels of tumour-infiltrating lymphocytes showed poor prognosis.100 101 Furthermore, the antitumour ability of NK cells has been shown to be reduced in patients with HCC.102 103 A study using an animal model demonstrated that the depletion of NK cells resulted in a decrease in the antitumour activity of IL-18/IL-12 therapy.104 With regard to the relationship between NK cells and alcohol use, many studies have reported an association between alcohol consumption and NK cell dysfunction. The NK cells in the peripheral blood of patients with alcoholic liver cirrhosis showed reduced cytotoxic activity against cancer cells.105 In an animal model, alcohol ingestion reduced the cytotoxicity of NK cells, which resulted from the reduction of NKG2D.9898 Furthermore, a recent study showed that alcohol consumption reduced the number of cytotoxic NK cells defined as Eomes+CD3−NK1.1+ in the liver.106 However, further studies are needed as it is difficult to completely clarify the association between NK cells and alcohol-induced HCC.
TLR4 and hepatocarcinogenesis in alcohol users
TLR4 recognises the lipid A motif of LPS, which is a component of Gram-negative bacteria. The plasma LPS levels were elevated in ethanol-fed animals and the plasma endotoxin levels were found to be elevated in patients with ALD.107 108 Machida et al109 reported that an HCV-derived protein, NS5A, induced the expression of the TLR4 gene in hepatocytes and B cells.109 They also showed that alcohol induced the progression of HCC through LPS-TLR4signaling activation. The LPS-TLR4 pathway was regulated by the Nanog-mediatedmodulation of the mitochondrial metabolism in NS5A Tg mice.110 These studies suggest the crucial involvement of TLR4 and NANOG in the induction of HCC that is mediated by alcohol and HCV infection.
Gene polymorphisms and HCC development in ALD
Several studies have demonstrated that genetic susceptibility is associated with the alcohol-induced cancer risk. There are several genetic polymorphisms that affect the clinical course of ALD as they can induce psychological behavioural changes, alcohol metabolism, lipid metabolism, oxidative stress-related pathway activation and inflammatory responses.69 As described above in the Alcohol absorption and metabolism section, the gene polymorphisms of ADH2 and ALDH2 have been reported to correlate with HCC development in a Japanese cohort study,21 111 but several studies conversely found no association with HCC69; thus, further validation is needed. Concerning alcohol-induced HCC development, the gene polymorphism of patatin-like phospholipase 3 domain containing 3 (PNPLA3) was found to be an important risk factor. Several cohort studies revealed the risk of PNPLA3 (rs738409) on alcohol-induced HCC in patients with ALD.112–114 A recent genome-wide association study in Europe revealed that the gene polymorphisms of TM6SF2 and PNPLA3 could be regarded as potential genetic risk factors for the development of alcohol-related HCC.115
Conclusion
Several clinical factors increase the risk of alcohol-induced HCC. A large alcohol intake, coexistence of diabetes and hepatitis virus infection, and female gender are established factors. The precise mechanisms of hepatocarcinogenesis are not clearly understood; however, there is increasing evidence to suggest that many factors are involved. Alcohol metabolites and adducts have been shown to induce oxidative stress, direct mutagenesis, and the aberrant methylation of DNA or protein on hepatocytes. Furthermore, the immune system is also implicated in the development and progression of HCC. Obviously, the best approach for resolving this complex pathogenesis is the cessation of alcohol consumption. However, as this is often difficult, further studies are needed to reduce the risk of HCC.
Acknowledgments
The authors wish to acknowledge Dr Hiroyuki Okada, Professor at the Department of Gastroenterology and Hepatology, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama, Japan.
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
Footnotes
Contributors HM and AT contributed to the writing of the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.