Article Text

Abstract
Background The role of Mycobacteriumavium subspecies paratuberculosis (MAP) in Crohn’s disease (CD) is increasingly accepted as evident by detection of the bacteria in the blood and intestinal tissue from patients with CD, and by supporting data from several open-label anti-MAP treatment studies. Tumour necrosis factor alpha (TNFα) monoclonal antibodies (anti-TNFα) have been widely used for CD treatment. Despite the short-term benefit of anti-TNFα in controlling CD symptoms, most patients suffer from detrimental adverse effects, including higher susceptibility to mycobacterial infections. Methods We investigated the effect of recombinant cytokines and anti-TNFα therapeutics on macrophages infected with clinical MAP strain isolated from CD patient blood. MAP viability was measured in macrophages pulsed with PEGylated and non-PEGylated anti-TNFα monoclonal antibodies at concentrations 0 to 50 µg/mL and with rTNFα, rIL-6, rIL-12, rIL-23 and IFNγ at a final concentration of 1000 U/mL. Expression of proinflammatory cytokines was measured by RT-PCR following MAP infection.
Results Both PEGylated and non-PEGylated forms of anti-TNFα increased MAP viability by nearly 1.5 logs. rIL-6 and rIL-12 induced MAP viability at 5.42±0.25 and 4.79±0.14 log CFU/mL, respectively. In contrast, rTNFα reduced MAP survival in infected macrophages by 2.63 logs. Expression of TNFα, IL-6 and IL-12 was upregulated threefold following MAP or M. tuberculosis infection compared with other bacterial strains (p<0.05), while expression of IL-23 and IFNγ was not significant after MAP infection.
Conclusion The data indicate MAP-positive patients with CD receiving anti-TNFα treatment could result in favourable conditions for MAP infection, which explains the poor response of many patients with CD to anti-TNFα therapy.
- TNF-Alpha
- crohn’s disease
- mycobacterium paratuberculosis
- inflammatory bowel disease
- cytokines
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See:©http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Summary box
What is already known about this subject?
Mycobacterium avium subspecies paratuberculosis (MAP) is among the most investigated pathogens associated with Crohn’s disease (CD).
Although the standard CD therapy has shifted from general immunosuppressants towards more specific targets including tumour necrosis factor alpha (TNFα), patients are often unable to reach full remission to this treatment.
Antagonizing TNFα have several adverse effects, including higher risk for multiple infections especially mycobacterial infections such as Mycobacterium tuberculosis, a closely related micro-organism to MAP.
What are the new findings?
Anti-TNFα therapeutics unexpectedly demonstrated similar cytotoxic effects to recombinant TNFα on uninfected macrophages.
Anti-TNFα therapeutics induce MAP survival in infected macrophages, which is a serious issue for CD patients who have MAP infection.
MAP induces expression of TNFα, IL-6 and IL-12 in infected macrophages.
Unlike rTNFα, rIL-6 induced MAP survival in infected macrophages, suggesting that IL-6 in MAP-infected patients with CD might be a better drug target.
Summary box
How might it impact on clinical practice in the foreseeable future?
MAP is accepted to be a serious pathogen in some patients with CD.
We have demonstrated that TNFα is the essential cytokine required for containment and eradication of MAP. It is, therefore, alarming for patients with CD who are infected with MAP to receive anti-TNFα therapeutics. Alternative CD therapeutic drug targets should be considered instead.
Higher expression of TNFα, IL-6 and IL-12 following MAP infection in vitro shows that the elevated level of these cytokines in patients with CD might be due to MAP infection.
Eradicating MAP infection with anti-MAP antibiotics is highly expected to suppress expression of proinflammatory cytokines.
The positive effects of recombinant IL-6 and IL-12 on MAP survival (unlike TNFα) directs the attention to novel therapeutic targets with dual effects on suppressing the immune response and MAP infection.
Introduction
Crohn’s disease (CD) is described as a complex idiopathic inflammation, which can affect any part of the digestive tract. Patients diagnosed with this chronic form of inflammatory bowel disease suffer from persistent diarrhoea, abdominal pain and malnutrition.1 The prevalence of CD in western countries increased recently, which carries a huge economic burden on healthcare cost, since 50% of patients with CD require surgical intervention within 10 years of diagnosis.1–3
The aetiology of CD involves various components including genetic susceptibility, altered microbiota and environmental triggers.4 One of the most investigated pathogens associated with CD is Mycobacterium avium subspecies paratuberculosis (MAP).5 6 However, current treatment guidelines do not consider this bacterial infection as a source of inflammation. Although the standard CD therapy has shifted from general immunosuppressants such as corticosteroids and thiopurines towards more specific targets including tumour necrosis factor alpha (TNFα), patients are usually unable to reach full remission or at least maintain a sustainable clinical response to this kind of therapeutic approach.7–9
Introducing anti-TNFα biologics (infliximab, adalimumab, certolizumab pegol) to CD treatment has shown that targeting a specific cytokine could be helpful to control CD symptoms and reduce flare-ups.7 Unfortunately, sustainable remission is very limited to a small group of patients.9 It has been reported that 10%–30% of patients with CD have no initial response to anti-TNFα therapeutics, and over 50% of initial responders lose their response to treatment over time.10 Additionally, about 40% of patients with CD are at risk of disease relapse after anti-TNFα treatment discontinuation.11
Moreover, blocking TNFα carries several adverse effects, including higher risk for malignancy, heart failure and multiple infections.4 There is a well-established evidence supporting the role of anti-TNFα therapeutics in increasing the incidence for mycobacterial infections including Mycobacterium tuberculosis, which is due to the importance of TNFα in granuloma formation and containment of M. tuberculosis.12 Since MAP and M. tuberculosis share molecular similarities and they both avoid phagosome–lysosome fusion in infected macrophages,13–15 TNFα remains the essential cytokine required for containment and eradication of MAP. It is, therefore, alarming for patients with CD who are infected with MAP to receive anti-TNFα therapeutics, and an extensive search for alternative CD therapeutic drug targets is required. Nevertheless, anti-TNFα treatment might still be considered for patients with CD who do not have MAP infection and who are genetically more likely to respond to this treatment.16
For refractory cases of CD, there are a few current medications with a novel therapeutic pathway known as integrin inhibitors, such as natalizumab (Tysabri) and vedolizumab (Entyvio). Although these two medications have shown a clinical efficacy in CD treatment, however, they increased the risk for progressive multifocal leucoencephalopathy in multiple clinical trials.17–19 Besides, several proinflammatory cytokines are emerging as possible therapeutic targets for CD such as IL-6, IL-12 and IL-23.20 However, the effect of these medications on mycobacterial infection is unknown. Additionally, the effect of MAP infection on upregulating proinflammatory cytokines in CD needs further investigation.
In this study, we focused on elucidating the effects of non-PEGylated and PEGylated anti-TNFα monoclonal antibodies on MAP survival in infected human-derived macrophages. We also evaluated the ability of recombinant cytokines (TNFα, IL-6, IL-12, IL-23 and IFNγ) to modulate MAP survival in vitro. Finally, we evaluated the effects of MAP infection on the gene expression level of these proinflammatory cytokines.
Materials and methods
Effect of anti-TNFα monoclonal antibodies and recombinant TNFα on MAP in culture
BD Bactec MGIT Para-TB medium (Becton Dickinson, Sparks, Maryland, USA) system was used to determine the bactericidal effects of anti-TNFα therapeutics against clinical MAP strain (UCF4; isolated from intestinal biopsy of a patient with CD) and other micro-organisms as controls. MGIT-para media with supplements were used to culture mycobacteria as described previously.21 For other micro-organisms, nutrient broth media replaced MGIT-para media in MGIT tubes. All tubes contained a fluorescent molecule embedded in an oxygen-sensitive silicone, where fluorescence is detected in the presence of active respiring bacteria. Anti-TNFα therapeutics were tested in concentrations between 0 and 200 µg/mL. Recombinant TNFα (rTNFα) was obtained as sterile filtered lyophilised powder (Gemini), where each 1.0 µg of powder had 2500 U. Then, 50 µg was dissolved in 500 µL of sterile water in order to prepare 0.1 mg/mL stock solution, which was stored at −20°C. Then, rTNFα was added to bacterial cultures at final concentrations between 0 and 1000 U/mL. The micro-organisms included in this study are MAP UCF4, Listeria monocytogenes ATCC 19112, Klebsiella pneumoniae ATCC 13883 and recombinant Escherichia coli. All culture tubes were incubated in BD Bactec MGIT 320 Analyzer at 37°C. Mycobacterial cultures were read weekly whereas other micro-organism cultures were read daily. Bacterial growth was visualised under the UV by using Andromeda BioSens SC 645 UV illuminator.
Effect of anti-TNFα monoclonal antibodies and recombinant TNFα on THP-1 cell viability and measurement of apoptosis
THP-1 cells (ATCC TIB-202) were cultured in RPMI-1640 medium containing 9.8% fetal bovine serum (FBS) (Sigma Life Science) and 0.09% 2-mercaptoethanol (BME) (Gibco Life Technologies), while maintained in a humidified 5% CO2 incubator at 37°C. Cells were grown until confluency in tissue culture plates was reached. Then, cells were distributed in 1 mL aliquots in a 24-well tissue culture plate. THP-1 cells were then treated with recombinant TNFα or anti-TNFα monoclonal antibodies at final concentrations between 0 and 50 µg/mL, followed by incubation of 24, 48 and 72 hours. Cell viability was determined by trypan blue exclusion assay. Briefly, 10 µL of cell suspension was mixed with 10 µL of 0.4% trypan blue solution. Following 5 min of incubation at room temperature, 10 µL of mixture was injected on a haemocytometer, and the percentage of stained (viable) cells was counted by binocular microscope. The apoptotic activities of recombinant TNFα and anti-TNFα monoclonal antibodies were determined by caspase-3 colorimetric assay (Abcam). At each time point, 500 µL of each cell suspension was mixed with 50 µL of chilled lysis buffer and incubated on ice for 10 min. Mixture was centrifuged for 1 min at 10 000g. The cytosolic extract was transferred to another tube and protein concentration was measured and adjusted to 100 µg per 50 µL of samples. Caspase reaction mix was prepared by mixing 50 µL of reaction buffer with 0.5 µL of 1,4-dithiothreitol for each reaction in duplicates. Each sample was mixed with 50 µL of reaction buffer and 5 µL of 4 mM DEVD-p-NA substrate on a 96-well microplate. Following 90 min of incubation at 37°C, the output was measured at 405 nm wavelength by using a microplate reader. Fold increase in caspase-3 activity was determined by comparing treated samples with the untreated control.
Infection of monocyte-derived macrophages with MAP
THP-1 cells (ATCC TIB-202) were cultured as described earlier, then they were differentiated into monocyte-derived macrophages by adding 50 ng/mL of phorbol 12-myristate 13-acetate (PMA) (Sigma-Aldrich) for 24 hours of incubation. Monocyte-derived macrophages were then infected with 1×107 bacteria/mL. Micro-organisms used include viable M. tuberculosis HR237, MAP UCF4 and M. smegmatis ATCC 27199, and heat-inactivated M. tuberculosis HR237, MAP UCF4 and M. smegmatis ATCC 27199. Non-mycobacterial micro-organisms included L. monocytogenes ATCC 19112, K. pneumoniae ATCC 13883 and recombinant E. coli. All micro-organisms were used at a final concentration of 1×107 viable bacteria/mL, while maintained at the same conditions for 24 hours.
Measurement of TNFα, IL-6, IL-12, IL-23 and IFNγ expression in infected macrophages
RNA was isolated from 1.0 mL of each sample suspension after 24 hours of infection and used for cDNA synthesis in order to analyse expression of TNFα, IL-6, IL-12, IL-23 and IFNγ via RT-PCR. Briefly, cells were centrifuged at 100g for 5 min. Pellets were suspended in 1.0 mL of TRIzol reagent (Invitrogen) for 15 min and then mixed with 0.2 mL of chloroform. Following 3 min of incubation at room temperature, samples were then centrifuged at 12 100g for 15 min at 4°C. The RNA was transferred from the upper aqueous colourless part into a new 2.0 mL microcentrifuge tube. Each sample was mixed with 0.5 mL of 100% isopropanol following 10 min of incubation at room temperature. Then, each sample was centrifuged at 12 100g for 10 min at 4°C and mixed with 1.0 mL of 75% ethanol, followed by centrifugation at 7000g for 5 min at 4°C. After RNA pellets were air-dried for 15 min, samples were suspended in 20 µL of RNase-free water and finally heated at 60°C for 10 min.
A total amount of 600 ng of each RNA sample was added to 0.2 mL of PCR reaction, 4 µL of iScript Reverse Transcription (Bio-Rad) and up to 20 µL RNase-free water for cDNA synthesis. All samples were transferred to a thermal cycler (MyGene Series Peltier Thermal Cycler), where reaction was performed for 5 min at 25°C, 20 min at 46°C and 1 min at 95°C. A total volume of 1 µL of cDNA (30 ng/µL) was mixed with 10 µL of Fast SYBR Green Mastermix (Thermo Fisher Scientific), 1 µL of either IL-6, IL-12, IL-23 or IFNγ PrimePCR SYBR Green Assay mix (Bio-Rad), in addition to 8 µL of molecular biological grade sterile water in a 96-well microamp RT-PCR reaction plate. The 18s RNA gene oligonucleotide primers (forward primer: 5'-GTA ACC CGT TGA ACC CCA TT-3'; reverse primer: 5'-CCA TCC AAT CGG TAG TAG CG-3') were used as controls to get baseline CT values. The RT-PCR reaction was performed using 7500 Fast Real-Time PCR System (Applied Biosystems). Relative mRNA expression levels were calculated by using the equation (2(−∆CT)×1000), where ∆CT=sample RT-PCR CT value−18s CT baseline value.
Modulating MAP viability in infected macrophages pulsed with exogenous recombinant cytokines and anti-TNFα monoclonal antibodies
Recombinant cytokines (rTNFα, rIL-6, rIL-12, rIL-23 and rIFNγ) were obtained as sterile filtered lyophilised powders (Gemini), where each 1.0 µg of powder had 2500 U. Then, 50 µg of each powder was dissolved in 500 µL of sterile water in order to prepare 0.1 mg/mL stock solution, which was stored at −20°C. Recombinant cytokines at final concentrations between 0 and 1000 U/mL were added to monocyte-derived macrophages infected with MAP strain UCF4. Similarly, two anti-TNFα monoclonal antibodies (PEGylated and non- PEGylated forms) at final concentrations between 0 and 50 µg/mL were also evaluated. Then, MAP viability was tested after 24, 48 and 72 hours.
Measurement of bacterial viability in infected macrophages
After infecting monocyte-derived macrophages with MAP for 24 hours, cells were washed to remove extracellular bacilli, and then recombinant cytokines/anti-TNFα monoclonal antibodies were added to the culture accordingly. Monocyte-derived macrophages were collected at three time points (24, 48 and 72 hours) and lysed with 200 µL of M-PER Mammalian reagent (Thermo Scientific) for bacterial viability detection using Live/Dead Baclight (Thermo Fisher Scientific). Briefly, five different proportions of live and dead MAP were prepared (0:100, 10:90, 50:50, 90:10, 100:0), then 100 µL of each bacterial suspension was pipetted in triplicates into separate wells of 96-well flat-bottom microplate and mixed 100 µL of staining reagent mixture. Following 15 min of incubation at room temperature in the dark, fluorescence intensity was measured at 530 nm, which indicates live bacteria reading (green), and again at 630 nm for the dead bacteria reading (red). Data were analysed by dividing the fluorescence intensity of the stained bacterial suspensions (ratio=emission 1 (green)/emission 2 (red)). The least-squares fit line was generated and the equation was used to calculate the bacterial viability from the infected cells post-treatment.
Statistical analysis
Data are expressed as mean±SE of the mean by using GraphPad Prism V.7.02 (GraphPad, La Jolla, California, USA). Two-tailed Student’s t-test was used. The difference between samples and controls was considered statistically significant at a level of p <0.05.
Results
Anti-TNFα therapeutics and recombinant TNFα have no direct bactericidal activity against MAP or other tested micro-organisms
In order to rule out the direct bactericidal effects of anti-TNFα monoclonal antibodies and rTNFα, we tested them in supratherapeutic levels against MAP and non-mycobacterial MGIT cultures. Both PEGylated and non-PEGylated anti-TNFα monoclonal antibodies have demonstrated no direct bactericidal activity when they were tested against MAP in addition to three non-mycobacteria strains (L. monocytogenes ATCC 19112, K. pneumoniae ATCC 13883 and recombinant E. coli). All bacterial strains we tested showed resistance to anti-TNFα treatment at concentrations as high as 200 µg/mL (4× Cmax). Similarly, all strains we tested were resistant to rTNFα treatment at concentrations as high as 1000 U/mL. Figure 1 shows the growth florescence of MGIT tubes incubated with MAP after 14 days of exposure to anti-TNFα, while figure 2 shows the growth fluorescence of MGIT tubes incubated with non-mycobacteria strains following 72 hours of anti-TNFα exposure.

Direct bactericidal activity of non-PEGylated anti-TNFα (1) and PEGylated anti-TNFα (2) therapeutics against MAP growth in MGIT fluorescence system at 200 µg/mL, following 14 days of incubation. (P) indicates positive control (0 µg/mL) and (N) is a negative control.

Direct bactericidal activity of non-PEGylated anti-TNFα (A) and PEGylated anti-TNFα (B) therapeutics against bacterial growth in MGIT fluorescence system at 200 µg/mL, following 72 hours of incubation. (1): S. aureus, (2): recombinant E. coli, (3): K. pneumoniae and (4): L. monocytogenes. (P) indicates positive control (0 µg/mL) and (N) is a negative control.
Anti-TNFα therapeutics demonstrate similar cytotoxic effects to recombinant TNFα on uninfected macrophages
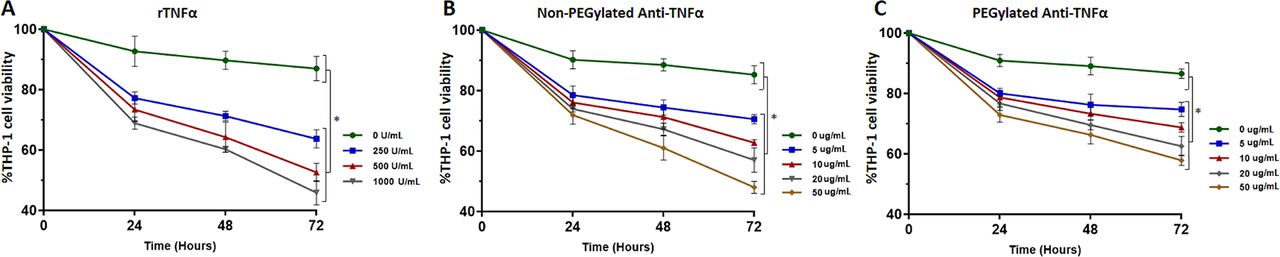
To further establish the cytotoxic role of TNFα and anti-TNFα monoclonal antibodies on monocyte apoptosis and whether these alone are able to modulate monocyte apoptosis independently of MAP infection, we treated uninfected THP-1 cells with different concentrations of exogenous rTNFα and therapeutic levels of anti-TNFα antibodies. Following 24 hours of incubation with 250 U/mL of rTNFα, THP-1 cell viability has decreased from 100% to 77.2%, which continued to decline over time until it reached 63.7% after 72 hours. In a concentration-dependent manner, 500 U/mL of rTNFα led to a decrease in THP-1 cell viability from 100% to 52.6%, while it reached 45.9% with 1000 U/mL, following 72 hours of incubation (p<0.05) (figure 3A). Caspase-3 activity has confirmed these findings since it has shown higher activity in THP-1 cells treated with rTNFα. At 250 U/mL, caspase-3 activity was 2.47-fold higher than control, while it was 6.49-fold and 8.92-fold higher in cells treated with 500 and 1000 U/mL of rTNFα following 72 hours of incubation, respectively (p<0.05) (figure 4A). Surprisingly, anti-TNFα monoclonal antibodies showed similar cytotoxic effects as rTNFα in a dose-dependent manner. The viability of THP-1 cells treated with 5 µg/mL of non-PEGylated anti-TNFα started to decrease over 72 hours of incubation, when it reached 70.6%. Higher concentrations have shown further decline in cell viability, when it reached 62.8%, 57% and 48% following 72 hours of incubation with 10, 20 and 50 ug/mL, respectively (p<0.05) (figure 3B). The PEGylated form of anti-TNFα was less cytotoxic, but it still demonstrated decline in THP-1 cell viability following 72 hours of incubation, when cell viability was 74.7%, 68.8%, 62.6% and 57.9%, treated with 5, 10, 20 and 50 µg/mL, respectively (p<0.05) (figure 3C). Similarly, caspase-3 activity was 4.9-fold and 4.4-fold higher than control following 72 hours of treatment with 50 µg/mL of non-PEGylated and PEGylated anti-TNFα therapeutics, respectively (p<0.05) (figure 4B,C).

Cytotoxicity of rTNFα (A), non-PEGylated anti-TNFα (B) and PEGylated anti-TNFα (C) therapeutics determined by Trypan blue exclusion assay, following 24, 48 and 72 hours of incubation. *P<0.05.

Cytotoxicity of rTNFα (A), non-PEGylated anti-TNFα (B) and PEGylated anti-TNFα (C) therapeutics determined by caspase-3 activity assay, following 24, 48 and 72 hours of incubation. *P<0.05.
MAP induces expression of TNFα, IL-6 and IL-12 in infected macrophages
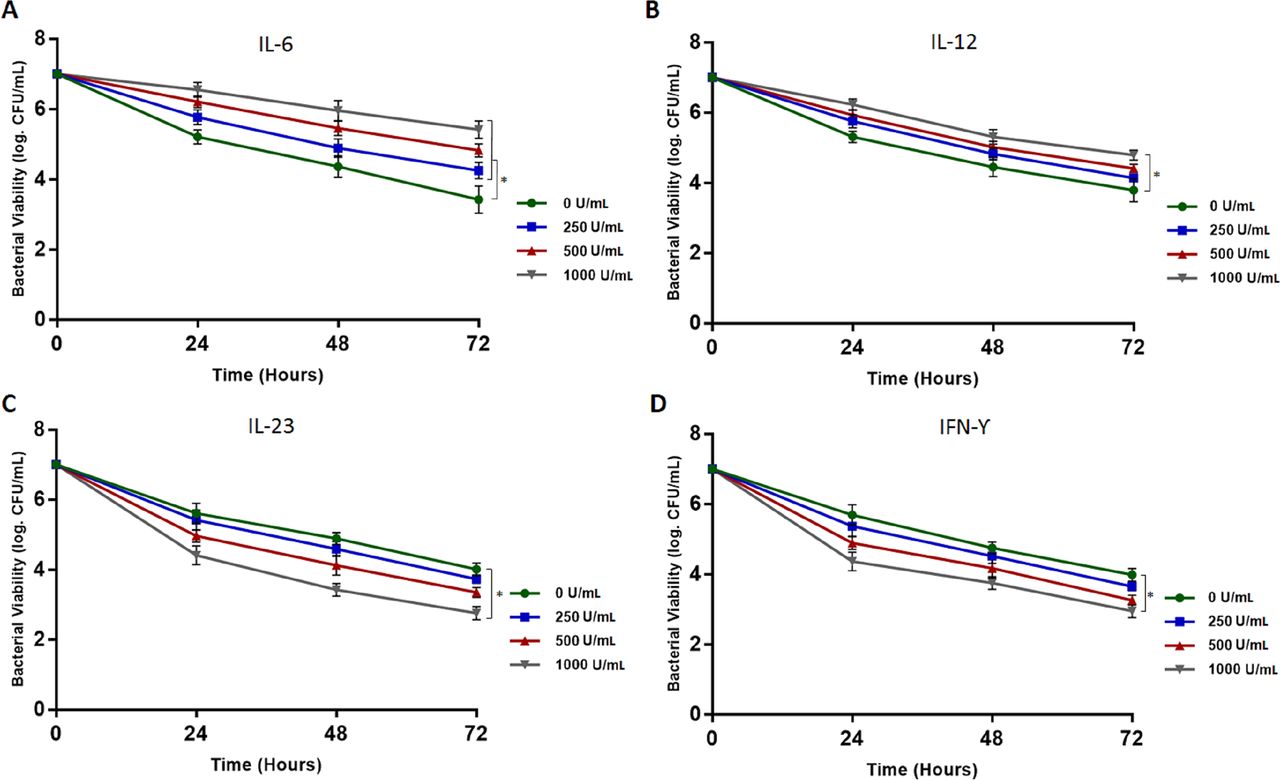
We have found that MAP and M. tuberculosis induce TNFα expression in infected macrophages significantly (3.2±0.23 and 2.8±0.13, respectively) compared with M. smegmatis (0.8±0.17) and other non-mycobacteria species following 24 hours of infection (figure 5A). Similarly, we found that MAP and M. tuberculosis both had about two times higher IL-6 expression (2.9±0.41 and 2.6±0.19, respectively) compared with M. smegmatis (1.3±0.12) or other non-mycobacterial pathogens (p<0.05). Furthermore, when M. tuberculosis and MAP were both heat inactivated (80°C for 20 min), they have lost their activity to induce IL-6 expression (0.35±0.14 and 0.29±0.11, respectively) (p<0.05) (figure 5B). Additionally, IL-12 expression was also significantly higher in MAP or M. tuberculosis infected macrophages (1.6±0.36 and 1.5±0.29, respectively), but only in comparison to non-mycobacteria species (p<0.05) (figure 5C). However, the expression of other proinflammatory cytokines (IFNγ and IL-23) was not significantly higher in MAP or M. tuberculosis infected macrophages (figure 5D,E).

Expression of TNFα (A), IL-6 (B), IL-12 (C), IL-23 (D) and IFNγ (E) in monocyte-derived macrophages following 24 hours of bacterial infection. Bacterial strains presented in the X-axis are 1: control (without infection), 2: M. tuberculosis (heat inactivated), 3: MAP UCF4 (heat inactivated), 4: M. tuberculosis, 5: MAP UCF4, 6: M. smegmatis, 7: K. pneumoniae, 8: L. monocytogenes, 9: recombinant E. coli. *P<0.05.
Recombinant TNFα decreases MAP survival in infected macrophages
First, we tested the effects of adding exogenous rTNFα into MAP infected macrophages at different concentrations between 0 and 1000 U/mL. We found that MAP viability drops with time significantly in a concentration-dependent manner. At 500 U/mL, MAP viability has declined from 7.0 (log CFU/mL) to 3.95±0.22, 3.31±0.34 and 2.11±0.17, following 24, 48 and 72 hours of incubation, respectively (p<0.05) (figure 6A). At the maximum concentration we have tested, adding rTNFα at 1000 U/mL to MAP infected macrophages had decreased MAP viability from 7.0 (log CFU/mL) to 1.50±0.15, following 72 hours of incubation (p<0.05) (figure 6A).
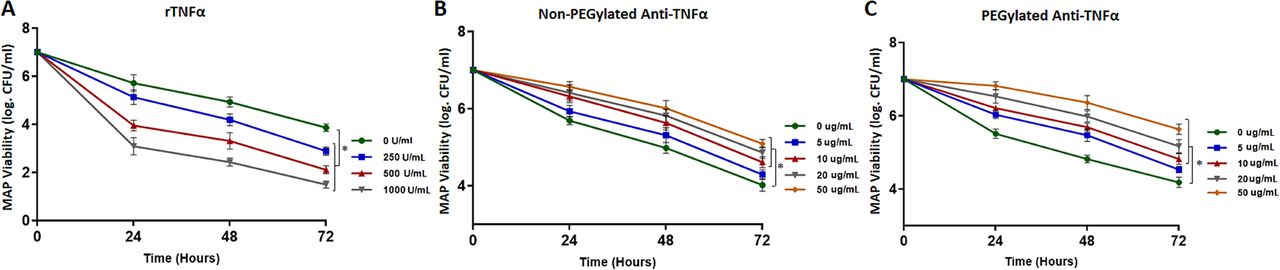
Mycobacterium avium subspecies paratuberculosis (MAP) viability in monocyte-derived infected macrophages pulsed with rTNFα (A), non-PEGylated anti-TNFα (B) and PEGylated anti-TNFα (C) therapeutics, following 24, 48 and 72 hours of infection. *P<0.05.
Anti-TNFα therapeutics increase MAP survival in infected macrophages
In contrast, treating MAP infected macrophages with anti-TNFα monoclonal antibodies increases MAP survival in a dose-dependent manner. The non-PEGylated form of anti-TNFα at 50 µg/mL had MAP viability of 5.09±0.11 (log CFU/mL), while the untreated control had 4.02±0.16 (log CFU/mL) (p<0.05) (figure 6B). Similarly, the PEGylated anti-TNFα had increased MAP viability up to 5.63±0.14 (log CFU/mL) at 50 µg/mL following 72 hours of incubation (p<0.05) (figure 6C).
Recombinant IL-6 promotes MAP survival in infected macrophages
Since we found that MAP modulates some proinflammatory cytokine gene expression in infected macrophages, it was worthy to test the effects of exogenous recombinant cytokines on MAP survival. We tested rIL-6 at different concentrations between 0 and 1000 U/mL. Interestingly, rIL-6 has promoted MAP survival significantly in a concentration-dependent manner. Following 72 hours of incubation, MAP viability was 3.42±0.39, 4.25±0.23, 4.82±0.19 and 5.42±0.25 (log CFU/mL), at 0, 250, 500 and 1000 U/mL of rIL-6, respectively (p<0.05) (figure 7A). Similarly, rIL-12 had some positive effect on MAP viability in infected macrophages; however, it was only significant at a concentration of 1000 U/mL, when MAP viability was 4.79±0.14 compared with 3.73±0.33 (log CFU/mL), following 72 hours of incubation (p<0.05) (figure 7B). On the other hand, both rIL-23 and rIFNγ had similar effects to rTNFα, where they had detrimental effects on MAP survival. MAP viability has declined from 7.0 to 2.76±0.16 and 2.95±0.19 (log CFU/mL), following 72 hours of incubation with 1000 U/mL of rIL-23 and rIFNγ, respectively (figure 7C,D).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
MAP viability in monocyte-derived infected macrophages pulsed with recombinant IL-6 (A), IL-12 (B), IL-23 (C) and IFN-γ (D), following 24, 48 and 72 hours of infection. *P<0.05.
Discussion
Macrophages exhibit several mechanisms in order to eradicate intracellular pathogens, such as releasing reactive oxygen intermediates, changing phagolysosomal acidity and production of proinflammatory cytokines.22 TNFα is one of those cytokines, which has a prominent role during mycobacterial infection, resulting in a formation of a complex circuit of other cytokines able to modulate T cells and macrophages response to infection and granuloma formation.23 Consequently, blocking TNFα function in animal models induced mycobacterial proliferation and reduced granuloma formation, indicating that TNFα is a primary cytokine for protection against mycobacterial infection.24–26 However, current treatment guidelines for many inflammatory disorders of supposed non-infectious origin recommend blocking TNFα pathway in order to suppress the hyperactive immune response and inflammation.27
Although there is a strong evidence demonstrating the involvement of MAP in CD, there is no recommendation of using antibiotics for CD treatment so far.21 On the contrary, the clinical use of anti-TNFα therapeutics has increased the risk for multiple infections including tuberculosis.28–30 Thus, assessment of latent tuberculosis infection status is highly recommended in order to determine if any patient intended to initiate anti-TNFα therapy has a risk for development of active disease.31
This study was concerned with identifying the detrimental ability of recombinant TNFα, IL-6, IL-12, IL-23 and IFNγ on MAP survival in infected macrophages and if blocking TNFα function by anti-TNFα monoclonal antibodies modulates MAP viability in vitro. Additionally, our goal was to identify which proinflammatory cytokines are highly expressed by macrophages following MAP infection. A recent study has reported that infliximab treatment increases MAP viability in infected macrophages from patients with CD by predominant induction of MAP dormant form.32 Our data show that both PEGylated and non-PEGylated forms of anti-TNFα therapeutics do not have any direct bactericidal effects against MAP or other non-mycobacterial strains at supratherapeutic concentrations (>200 mg/mL). However, these medications increased MAP survival in infected macrophages in a dose-dependent manner, which indicates that patients with CD receiving such treatment are at a higher risk for MAP growth if they had MAP infection before initiation of therapy. In contrast, MAP viability declined in infected macrophages pulsed with exogenous rTNFα in a concentration-dependent manner, which shows that TNFα plays a significant role in protection against MAP infection.
Furthermore, anti-TNFα therapeutics demonstrated similar cytotoxicity level to rTNFα at therapeutic concentrations, which explains why these medications increase the risk for infections once they induce apoptosis in macrophages. In addition, we measured expression level of TNFα, IL-6, IL-12, IL-23 and IFNγ in infected macrophages following 24 hours. We found that TNFα, IL-6 and IL-12 are also expressed significantly in MAP or M. tuberculosis infected macrophages, which shows that a high level of these cytokines in patients with CD could be a result of MAP infection. Interestingly, MAP survival was induced significantly when exogenous rIL-6 was added to infected macrophages in a concentration-dependent manner. However, rIL-23 and IFNγ had a similar effect to rTNFα, where they reduced MAP viability significantly with higher concentrations.
Newly emerging monoclonal antibodies indicated for CD treatment have shifted from targeting TNFα into more selective targets such as anti-IL-6 (PF-04236921), anti-IL-23 (AMG-139) and anti-IL-12/IL-23 (ustekinumab).20 Indeed, IL-6 is highly expressed in patients with CD.33–35 Therefore, blocking IL-6 pathway is anticipated to reduce the hyperactive immune response among patients with CD. Moreover, our data suggest that IL-6 promotes MAP survival in infected macrophages. Thus, specifically targeting this cytokine will lead to a decline in MAP viability, which could replace anti-TNFα treatment eventually. Additionally, in M. tuberculosis infected macrophages, IL-6 was found to inhibit IFNγ responsive genes at the transcriptional level selectively, which also results in inhibition of major histocompatibility complex (MHC) class II induction.36 Clinical studies are needed to offer a proof of principle for this new CD drug target with dual effect on MAP infection and inflammation.
Acknowledgments
Our thanks are due to Southlake Gastroenterology for providing us with anti-TNFα medications and all members of SAN’s laboratory.
References
Footnotes
Contributors AQ is the primary author who performed all experiments, collected data and participated in writing the manuscript. SAN is the leading investigator in the laboratory and has supervised all aspects of the study including writing and editing of the manuscript.
Funding This study was funded in part by the Florida Legislative Grant and UCF Doctoral Research Support Award.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.