Article Text

Abstract
Objectives Gut microbiota modifications occurring during HIV infection have recently been associated with inflammation and microbial translocation. However, discrepancies between studies justified a comprehensive analysis performed on a large sample size.
Design and methods In a case–control study, next-generation sequencing of the 16S rRNA gene was applied to the faecal microbiota of 31 HIV-infected patients, of whom 18 were treated with antiretroviral treatment (ART), compared with 27 healthy controls. 21 sera samples from HIV-infected patients and 7 sera samples from control participants were used to test the presence of 25 markers of inflammation and/or immune activation.
Results Diversity was significantly reduced in HIV individuals when compared with controls and was not restored in the ART group. The relative abundance of several members of Ruminococcaceae such as Faecalibacterium prausnitzii was critically less abundant in the HIV-infected group and inversely correlated with inflammation/immune activation markers. Members of Enterobacteriaceae and Enterococcaceae were found to be enriched and positively correlated with these markers. There were significantly more aerotolerant species enriched in HIV samples (42/52 species, 80.8%) when compared with the control group (14/87 species, 16.1%; χ2 test, p<10−5, conditional maximum-likelihood estimate (CMLE) OR=21.9).
Conclusions Imbalance between aerobic and anaerobic flora observed in HIV faecal microbiota could be a consequence of the gut impairment classically observed in HIV infection via the production of oxygen. Overgrowth of proinflammatory aerobic species during HIV infection raises the question of antioxidant supplementation, such as vitamin C, E or N-acetylcysteine.
- HIV/AIDS
- OXIDATIVE STRESS
- INTESTINAL BACTERIA
- INFLAMMATION
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Summary box
What is already known about this subject?
▸ Gut microbiota disruptions occurring during HIV infection are associated with inflammation, immune activation or microbial translocation.
▸ Enterobacteriaceae and Prevotella spp. are frequently found to be enriched in HIV participants, whereas a decrease in Bacteroides spp. is commonly reported.
▸ Bacterial diversity of HIV gut microbiota is still debated, as well as the impact of antiretroviral treatment (ART) on microbial community restoration.
What are the new findings?
▸ Diversity was significantly reduced in HIV individuals and was not restored by ART.
▸ We found significantly more aerotolerant species enriched in HIV samples when compared with the control group.
▸ Species intolerant to oxygen such as Faecalibacterium prausnitsii or members of the Ruminococcus genus were depleted.
▸ Bacteria positively associated with inflammation were tolerant to oxygen, whereas those negatively associated were strictly anaerobic.
How might it impact on clinical practice in the foreseeable future?
▸ Markers of oxidative stress and gut redox potential could help to assess disease progression.
▸ Imbalance between aerobic and anaerobic communities in HIV participants raises the question of antioxidant supplementation, such as vitamin C, E or N-acetylcysteine.
Introduction
Studies concerning human gut microbiota are continuously growing,1 and numerous associations with human health and disease have been suggested, including inflammatory bowel diseases, energy metabolism or susceptibility to Clostridium difficile infections.2–9 The number of publications concerning HIV and gut microbiota was previously fairly modest but has also begun to increase in recent years. Indeed, although there is strong evidence of extensive structural changes in the relative abundance of microorganisms in the HIV faecal bacterial community, significant differences in regard to the sampled material or the platforms used for taxonomic assignment lead to heterogeneous conclusions. This has been particularly reflected in discrepancies concerning gut microbiota diversity during HIV infection, which were increased in some studies10 and depleted in others,11 ,12 while stomach fluid flora diversity has been described as reduced.13 Alongside these modifications, gut microbiota from US participants infected with HIV-1 have also been described as Prevotella-rich or Bacteroides-poor, similar to HIV-negative individuals in agrarian societies and in participants with high carbohydrate, low protein and low fat foods,10 suggesting an interplay with diet. Increase in the Prevotella community has nevertheless been linked with increased numbers of activated colonic T cells and myeloid dendritic cells (mDC).14 Moreover, focusing on colonic mucosal-adherent bacterial microbiota, depletion of Bacteroidales and an increase in Proteobacteria has been reported in HIV rectal biopsies.15 Interestingly, these disruptions have been associated with acceleration of tryptophan degradation through the kynurenine pathway.15 This has been positively correlated with gut impairment markers as well as levels of inflammatory cytokines.16 In addition, proinflammatory states and disease progression in HIV patients have been linked to gut bacterial community disruptions during HIV infection. For example, the high production of interleukin (IL)-6 and increased tumour necrosis factor (TNF)/IL-10 ratio have been associated with increased Enterobacteriaceae15 ,17 and Eubacterium biforme,18 respectively, and TCD8+ cell activation was mediated by Bacteroidales.19 While antiretroviral treatment (ART) appears to be able to reduce some abnormalities,12 it alone cannot consistently restore the gut microbiota of HIV participants to their native state.10 ,16 ,18 This is supported by the increase in genes involved in lipopolysaccharide (LPS) biosynthesis, or inflammatory pathways revealed by functional gene content sequencing of gut mucosa from patients receiving ART.20 Gut microbiota disturbances occurring during HIV infection may alter the functional role of the epithelial barrier towards pathogenic microorganisms.21–23 Finally, depletion of anaerobic prokaryotes was recently associated with microbiota of severe acute malnutrition,24 which shares several clinical similarities with the wasting syndrome, a progressive weight loss associated with AIDS, which has not disappeared despite the advent of ART.25
Accordingly, this study aimed to assess bacterial diversity and identify a taxonomic signature of HIV gut microbiota, including balance between aerobic species and those intolerant to oxygen. Secondarily, potential associations between enriched or depleted taxons with disease progression markers (CD4 T-cell count, viral load), inflammation markers and translocation markers were tested.
Materials and methods
Patients
In a case–control study following the STROBE statement,26 we compared the gut microbiota of 32 HIV-positive patients to that of 27 non-HIV healthy participants. HIV patients were recruited within the Infectious Diseases Departments in Conception and North Hospitals, Marseille, France, between 2012 and 2014. Non-HIV healthy controls were recruited during the same period and city using a snowball approach. Patients were eligible if they were adults (>18 years), HIV seropositive and provided a stool sample. Data collected included age, sex, antibiotic treatment within the three previous months, weight and height. In addition, we collected for cases only: CD4+ T cells, viral load and ART (see online supplementary table S1). Exclusion criteria included antibiotic treatment within the three previous months and body mass index <18.5 or ≥25 kg/m2. Signed written consent forms were obtained for all participants. Permission from the local ethics committee of the IFR48 (Marseille, France) was obtained under agreement 09-022 and ANRS EP55 MICROGUT.
supplementary data
Variables
Measured variables included gut bacterial relative abundances at the family, genus and species levels, presence of ART, CD4 T-cell count, viral load and seric concentrations of inflammation and translocation markers. HIV serology was determined with the Architect assay (Abbott Diagnostics, Mannheim, Germany). Plasma HIV RNA testing was performed using the RealTime HIV-1 assay (Abbott Diagnostics).
DNA extraction and sequencing
All participants' stool samples were stored at −80°C just after sample collection. Fifty-nine samples were extracted using a deglycosylation protocol, as follows: 250 µL of each sample was placed in a 2 mL tube containing a mixture of acid-washed glass beads (Sigma, Aldrich) and with two or three 0.5 mm glass beads. Mechanical lysis was performed by bead beating the mixture using a FastPrep BIO 101 apparatus (Qbiogene, Strasbourg, France) at maximum speed (6.5) for 3×30 s. The supernatant was centrifuged at 12 000 rpm for 10 min and the pellet retained. A mixture containing 2 µL of 10×glycoprotein denaturing buffer EndoHf (New England Biolabs) and 17 µL of H2O was added and heated at 100°C for 10 min. Deglycosylation was performed adding a mixture of 2 µL of 10×G5 reaction buffer (ref B1702 New England Biolabs), 2 µL of EndoHf (New England Biolabs), 2 µL of cellulase (Sigma) and 16 µL of H2O. The preparation was then incubated overnight at 37°C. Finally, DNA was extracted using the EZ1 biorobot (Qiagen) with the EZ1 DNA tissues kit. The elution volume was 50 µL.
Samples were amplified individually for the 16S ‘V3_V4’ regions by PCR on MiSeq technology using the Taq Phusion (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and the surrounding conserved regions V3_V4 primers with overhang adapters (FwOvAd_341F CGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG; RevOvAd_785RGTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC). After purification on AMPure beads (Beckman Coulter, Fullerton, California, USA), concentrations were measured using high sensitivity Qubit technology (Beckman Coulter) and dilutions to 0.2 ng/µL were performed. Using a subsequent limited cycle PCR on 1 ng of each PCR product, Illumina sequencing adapters and dual-index barcodes were added to each amplicon. After purification on AMPure beads (Beckman Coulter), libraries were then normalised according to the Nextera XT protocol (Illumina, San Diego, California, USA). The multiplexed samples were pooled into a single library for sequencing on the MiSeq (Illumina). Automated cluster generation and paired-end sequencing with dual index reads was performed in a single 39 hour run in a 2×250 bp. These samples were mixed with other amplicon projects, resulting in four runs being conducted. On average, a total of 7.86 Gb of information was obtained from a 1030 K/mm2 cluster density, with a cluster passing 74.48% quality control filters (21 587 600 clusters). Within these runs, the index representations for all samples were determined between 0.38% and 3.5%. Raw data were configured in fasta files for R1 and R2 reads as paired-end reads.
The paired-end reads from the Illumina Miseq raw fasta files were assembled into contigs using FLASH (Fast Length Adjustment of SHort reads),27 a tool to merge paired-end reads from next-generation sequencing experiments when the original DNA fragments are shorter than twice the length of reads. FLASH was used to clean the sequences at the same time of merging paired-end reads with a (OFFSET) quality score cut-off of 33. The longer joined sequences obtained from FLASH were filtered using QIIME,28 trimming off the primers and removing the sequences shorter than 200 nts and >1000 nts. The chimeric sequences were also removed using the QIIME chimeraslayer. Clustering of sequences into operational taxonomic units (OTUs) was performed by UCLUST29 and the QIIME de novo method at 97% similarity, without considering the singletons.
Building the reference database
The SILVA SSU and LSU database30 (release 119) was downloaded from the SILVA website and used to build a local database of predicted amplicon sequences. While building our local reference database, we only extracted SILVA SSU reference sequences if and only if they contained both the forward and reverse primers, allowing three differences between each primer and the SILVA reference sequences. Finally, our local reference database contained a total of 456 714 well-annotated sequences. One sample from an HIV participant was finally excluded from the study due to insufficient metagenomic data (<40 000 total high-quality filtered reads).
Taxonomic assignment
The OTUs (representative sequences) extracted in the previous step were blasted31 against our local reference database. The 100 best matches above 80% identity (100% coverage and e value of 0.000001) with each of the representative sequences were extracted from the reference database.
These extracted reference sequences with the SILVA taxonomy were sorted according to the decreasing percentage of similarity. The reference sequences with the highest percentage of similarity (also considering all the hits within a 0.5% similarity of the best hits) with the OTUs were then considered for taxonomic assignment. Taxonomy to the lowest rank was obtained by applying a majority voting.32 When similarity was 80%, sequences were not assigned. Finally, all the tags were clustered to different taxon ranks according to the consensus taxonomy of the unique tags (representative of each OTU).33–35 Rarefaction was not performed as it could lead to false-positive results.36 The details of sequence numbers and OTUs can be seen in online supplementary table S3. All the sequences in raw fastq format have been submitted to EMBL-EBI37 with the accession number PRJEB10578.
Disease progression markers and systemic inflammation profiles
Twenty-one sera samples from HIV-infected patients of which 15 under ART and 7 sera samples from non-HIV-infected participants were collected to test the presence of 25 markers of inflammation and/or immune activation. sCD14 and FABP-2/I-FABP were quantified using ELISA (Bio-Techne, Abington, UK). The other markers were quantified using Luminex multiplex bead-based technology and a Bio-Plex 200 instrument (Bio-Rad): IL-2Rα, IL-18, MIG/CXCL9 and TRAIL using Bio-plex Pro Human cytokine Group II (Bio-Rad, Marnes-La-Coquette, France); B7-H1/PD-L1, MIP-1α/CCL4, sCD27, sCD30, sCD40L, sCD163, CXCL16, interferon (IFN)-γ, IL-17A, IL-22, IL-18BPa, IL-6, IL-8/CXCL8, IL-10, IP-10/CXCL10, I-TAC/CXCL11, MCP-1/CCL2, TNF-α and MIP-3α/CCL20 using Human Magnetic Luminex screening Assay (Bio-Techne, Abington, UK) in line with the manufacturer's instructions. One replicate was performed for each assay.
Statistical analyses
Shannon diversity indices (H′) were calculated for each sample using the formula  .38 A subgroup analysis was planned a priori differentiating patients with and without ART. Statistical analyses were performed using GraphPad Prism, V.5.0 (La Jolla, California, USA). The Fisher's exact test was used for dichotomous variables, the Mann-Whitney test for continuous variables and the Spearman test for correlations. Principal coordinate analysis (PCoA) was obtained using the weighted unifrac distance after data rarefaction at the depth of 50 000 reads per sample, and the Adonis test was performed in QIIME. Linear discriminant analysis effect size (LEfSe) was performed on relative abundances at the genus and species levels using parameters recommended by Segata et al,39 including per-sample normalisation of the sum of the values to 1 M (http://huttenhower.sph.harvard.edu/galaxy/). Tolerance to oxygen for each species or genus was checked using the ‘List of Prokaryotes according to their Aerotolerant or Obligate Anaerobic Metabolism’ (http://www.mediterranee-infection.com/article.php?laref=374&titre=list-of-prokaryotes-according-to-their-aerotolerant-or-obligate-anaerobic-metabolism). Owing to the possible risk of missing important findings, adjustments for multiple comparisons were not performed, as suggested for exploratory work.40 However, in a sensitivity analysis to test the robustness of our results, we reanalysed our data set based on the relative abundance of each species after normalisation to 50 000 reads (samples with <50 000 16S rRNA reads were excluded, and all relative abundance inferior to 1/50 000 were converted to 0). A non-parametric Kruskal-Wallis test was thus used with an adjustment for multiple comparison using the post hoc Benjamini-Hochberg correction from the OMICS package in XLSTAT V.2016.02 (Addinsoft, Paris, France).
.38 A subgroup analysis was planned a priori differentiating patients with and without ART. Statistical analyses were performed using GraphPad Prism, V.5.0 (La Jolla, California, USA). The Fisher's exact test was used for dichotomous variables, the Mann-Whitney test for continuous variables and the Spearman test for correlations. Principal coordinate analysis (PCoA) was obtained using the weighted unifrac distance after data rarefaction at the depth of 50 000 reads per sample, and the Adonis test was performed in QIIME. Linear discriminant analysis effect size (LEfSe) was performed on relative abundances at the genus and species levels using parameters recommended by Segata et al,39 including per-sample normalisation of the sum of the values to 1 M (http://huttenhower.sph.harvard.edu/galaxy/). Tolerance to oxygen for each species or genus was checked using the ‘List of Prokaryotes according to their Aerotolerant or Obligate Anaerobic Metabolism’ (http://www.mediterranee-infection.com/article.php?laref=374&titre=list-of-prokaryotes-according-to-their-aerotolerant-or-obligate-anaerobic-metabolism). Owing to the possible risk of missing important findings, adjustments for multiple comparisons were not performed, as suggested for exploratory work.40 However, in a sensitivity analysis to test the robustness of our results, we reanalysed our data set based on the relative abundance of each species after normalisation to 50 000 reads (samples with <50 000 16S rRNA reads were excluded, and all relative abundance inferior to 1/50 000 were converted to 0). A non-parametric Kruskal-Wallis test was thus used with an adjustment for multiple comparison using the post hoc Benjamini-Hochberg correction from the OMICS package in XLSTAT V.2016.02 (Addinsoft, Paris, France).
Results
Participants
All variables had been assessed for the 59 participants (32 HIV and 27 healthy controls). However, one seropositive individual had been excluded because of the insufficient number of reads (<40 000; see online supplementary table S2).
Decreased diversity of gut microbiota from HIV participants
The α-diversity was significantly reduced in HIV individuals when compared with controls as shown by the lower Shannon index (H′=2931 and 3394, respectively; p=00 068). Diversity was not restored in the ART group when compared with the untreated group (H′=2974 and 2879, respectively; p=0.72; figure 1). However, data clustering analysed by PCoA using the weighted unifrac distance to obtain the PCoA in the QIIME pipeline shows marked separation between controls and HIV individuals (ADONIS test p=0.002), as between HIV-treated and HIV-untreated participants (ADONIS test, p=0.033; see online supplementary figure S1).

Shannon indices applied on HIV cases and non-HIV controls. ART, antiretroviral treatment.
Taxa discriminating HIV-negative and seropositive participants
All HIV individuals clearly show a significant decrease in the Clostridia class as compared with the control samples (p<0.0001; figure 2). Peptostreptococcaceae families (p=0.0216) and especially Ruminococcaceae (p<0.0001) were significantly decreased. More specifically, the genera Subdoligranulum (p=0.0002), Ruminococcus (p<0.0001), Blautia (p=0.0239) and Faecalibacterium (p=0.0354) were critically less abundant. At the species level, R. bromii (p<0.0001), R. obeum (p=0.0086), F. prausnitzii (p<0.0001), B. luti (p=0.0199) and B. faecis (p=0.0385) were decreased.

Cladogram yielding taxa enriched in each case and control group with an LDA score >2.
The Gammaproteobacteria class was significantly increased (p=0.0021) in HIV-infected participants (figure 2), including the Citrobacter genera (p=0.03) and Escherichia coli (p=0.0368).
Moreover, we observed a low relative abundance of Bifidobacterium genera (p=0.0458) and B. adolescentis (p=0.0111) at the species level in HIV-infected individuals, while the Enterococcus genus (p=0.0339), including E. faecalis (p=0.0248) and E. faecium (p=0.0376), were found to be enriched.
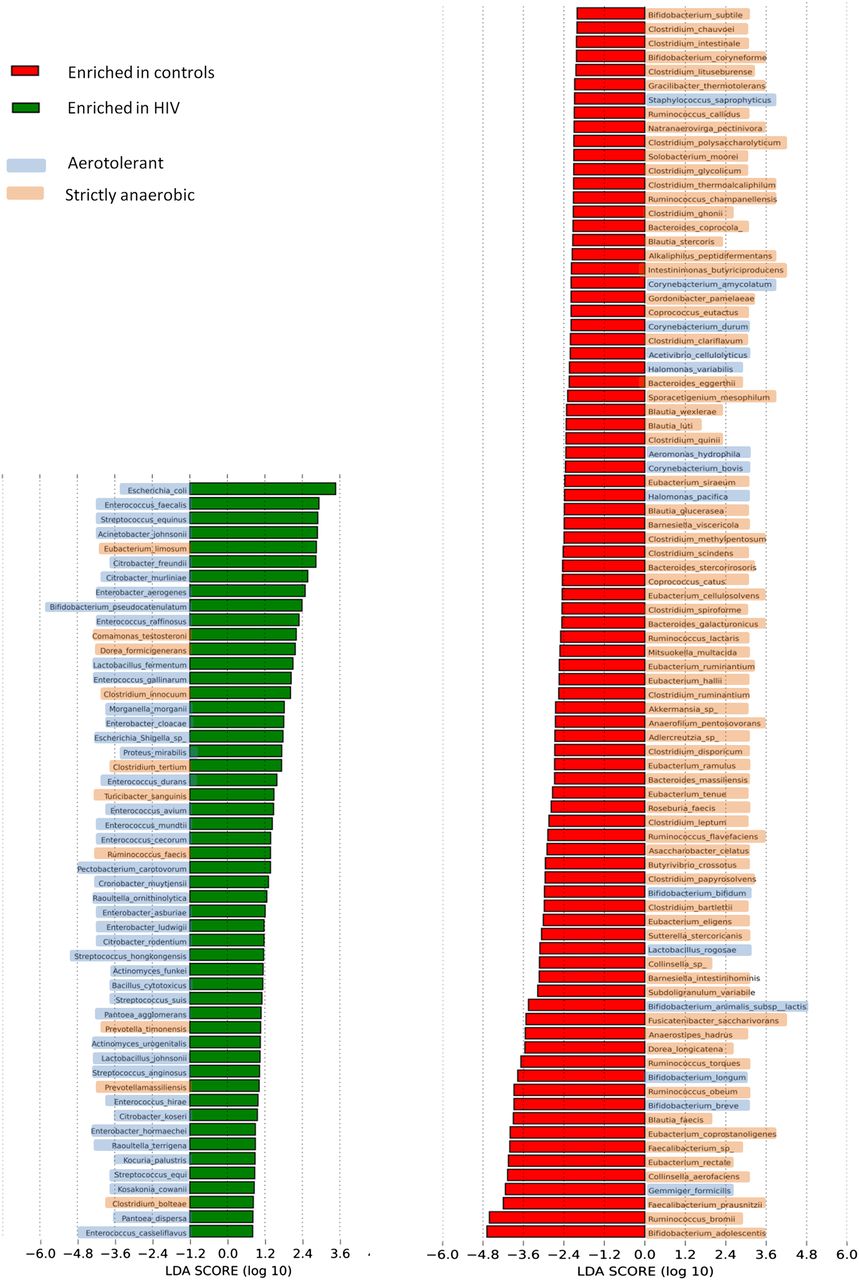
We also performed LEfSe at the species level to strengthen our results. LEfSe indeed confirmed enrichment of species belonging to Proteobacteria as diminution those belonging to Clostridia in HIV individuals (figure 2). Unprofitable taxa were removed and genera and species enriched in each group with an Linear Discriminant Analysis (LDA) score >2.0 were then kept. Interestingly, there were significantly more aerotolerant genera in the HIV group (12/18, 66.7%) than in the control group (5/29, 17.2%; χ2 test, p=4.8×10−4, CMLE OR=9.1; see online supplementary figure S2). Similarly, there were significantly more aerotolerant species enriched in HIV samples (42/52 species, 80.8%) when compared with the control group (14/87 species, 16.1%; χ2 test, p<10−5, conditional maximum-likelihood estimate (CMLE) OR=21.9; figure 3). Even though LDA is a validated method,38 we tested the robustness of our results performing an adjustment for multiple comparison (Benjamini-Hochberg, see the Materials and methods section) and normalisation to 50 000 reads. These secondary analyses confirmed our main result with a very significant depletion of anaerobic bacteria in HIV gut microbiota (CMLE OR=11.1, p=5.5×10−5; see online supplementary table S4).We then compared sequences from HIV individuals included in this study with those of uninfected controls from work by Dinh et al41 (project accession number SRP039076). Taxonomic assignment and LEfSe at the genus level were performed as described in the Materials and methods section. There were significantly more enriched aerotolerant genera in HIV included here (40/63, 63.5%) than in controls from the previously cited study (8/63, 12.7%; χ2 test, p<10−5, CMLE OR=11.7; see online supplementary figure S3).
{kind=link}
{kind=link}
{kind=link}
LDA scores of differentially abundant species among cases and controls and their tolerance to oxygen according to the ‘List of Prokaryotes according to their Aerotolerant or Obligate Anaerobic Metabolism’. The LDA scores represent the effect size of each abundant species. Species enriched in each group with an LDA score >2 are considered.
Disease progression markers and systemic inflammation profiles
Of the 25 markers studied, 7 (B7-H1/PD-L1, IFN-g, IL-17A, IL-22, IL-6, IL-10 and TNF-α) were below the lower limit of quantification and 12 were shown to be significantly different between HIV+ patients and HIV− participants using the Mann–Whitney rank test (see online supplementary figure S4). Chemokines and T-cell activation markers are correlated (see online supplementary figure S5), whereas no correlation was found between T-cell activation markers and translocation markers. These results suggested different mechanisms involved in the activation of T cells and monocytes/macrophages. We then looked at Spearman correlations between the relative abundance of bacteria families or species and levels of these biomarkers (table 1). Members of the Ruminococcaceae family (especially R. bromii and F. prausnitzii) were inversely correlated with inflammation/immune activation markers when depleted. Members of the Enterobacteriaceae family (especially E. coli and Enterobacter aerogenes) and of the Enterococcaceae family (E. faecalis and E. faecium) were positively correlated with these markers when enriched (table 1). Altogether, these results highlight that dysbiosis observed in HIV-infected patients is associated with systemic immune activation.
Significant associations found between different analysed markers (translocation markers, T-cell activation markers and chemokines) and specific taxa
Discussion
In this paper, we propose a metagenomic approach to stool samples collected from 31 HIV individuals and 27 controls. Identical special procedures and protocols were used from DNA extraction to amplicon analysis to ensure the robustness of our results.
First, our results found reduced gut microbiota diversity during HIV infection, revealed by lower Shannon indices in the HIV group independently of ART (figure 1). This point remains currently controversial with publications either confirming11 or contradicting this statement.10 A reduction in the bacterial diversity has recently been reported in the stomach fluid microflora of HIV-positive patients13 and this could be linked to bowel diseases.42 Alongside this modification, high structural changes had been observed in the HIV group, with a switch between species belonging to the Clostridia and those belonging to the Gammaproteobacteria classes, which were found to be, respectively, decreased and increased. Deep taxonomic analysis revealed a dramatic decrease in R. bromii, a keystone member of Ruminococcaceae for starch hydrolysis,43 in HIV patients. Interestingly, activation and maturation status of colonic mDC in HIV patients had been negatively associated with a low prevalence of mucosal species, including R. bromii.44 In addition, downregulation of starch metabolism had been observed in the intestinal epithelial barrier occurring during primary HIV infection.45 Besides, we found F. prausnitzii to be less abundant in HIV patients, which had been strongly associated with inflammatory bowel diseases such as Crohn's disease or colitis46–48 as the result of its anti-inflammatory properties which lead to its potential use as a probiotic.49 An increase in Gammaproteobacteria resulted from the high Enterobacteriaceae count, including the genera Citrobacter and Escherichia–Shigella and, more specifically, E. coli. Our findings are supported by a recent publication41 showing enrichment in Enterobacteriaceae, which are well known to be immune system activators and proinflammatory organisms through LPS mediation.17 Similar changes have also been observed in rectal biopsy microbiota from HIV participants.15 These disruptions are evidence of a deep imbalance between aerobic and anaerobic flora, which was still found when amplicon sequences from healthy participants41 from another study were considered as controls. The increase in oxygen within the gastrointestinal tract could be a consequence of the gut impairment classically observed in HIV infection, promoting facultative anaerobes. Interestingly, the increased bacteria associated with elevated activation immune markers were aerotolerant (table 1). In the same way, microorganisms for which depletion was correlated to the same activation immune markers were strictly anaerobic (5/5; table 1), and mainly concern the Ruminococcaceae family, in particular F. prausnitzii and R. bromii (table 1). To the best of our knowledge, this is the first report on the aerobic/anaerobic bacteria imbalance in HIV gut microbiota. However, it is the overall dysbiosis, possibly due to intestinal barrier impairment, which appears to be the cause of this proinflammatory state, rather than one particular species. These findings raise the question of the involvement of gut microbiota in oxidative stress occurring during HIV infection, as well as the possible introduction of vitamin C supplementation, which has been previously suggested to have a protective effect.50 ,51 In addition, in vitro studies demonstrated that supplementation with vitamin C and N-acetylcysteine in combination reduced early apoptosis of CD4+ T-cell activation induced by LPS.52 Moreover, selenium supplementation protects infected T cells against an H2O2 cytotoxic effect, increasing glutathione peroxidase and decreasing nuclear factor-κB activation.53 Unfortunately, the impact of antioxidants on gut microbiota composition has not been studied, to the best of our knowledge, even if their use in microbiology can promote growth of anaerobes in the presence of oxygen.54
We assume that the low number of concomitant sera collected (N=27) and the fact that diet had not been taken into account constitute limitations of this work. In addition, it would be interesting to sample mucosal specimens rather than faeces samples.
In conclusion, this study highlights the loss of gut microbiota diversity during HIV infection, associated with an increase in aerobic bacteria such as Enterobacteriaceae or Enterococcaceae, and the depletion of anaerobic bacteria, mainly represented by the Ruminococcaceae family. The association of these disruptions with several activation or translocation markers raises the question of gut bacterial community restoration as an adjuvant treatment for HIV infection.
Acknowledgments
The authors wish to thank Emile Foucat for his valuable help in assaying the markers analysed in this study.
References
Footnotes
Contributors DR designed the whole study, monitored data collection, as well as drafted and revised the paper. GD monitored the data collection, analysed the data, as well as drafted and revised the paper. J-CL initiated the collaborative project, as well as drafted and revised the paper. PB, AS, SM and IR monitored the sample collection and revised the draft paper. CR and CM monitored the metagenomics analysis, as well as drafted and revised the paper. DB analysed the metagenomics data, as well as drafted and revised the paper. MS, SH and YL performed the correlation markers analysis, as well as drafted and revised the paper. MM performed the statistical analysis and revised the paper.
Funding This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Ethics approval Approval from local ethics committee of the IFR48 (Marseille, France) was obtained under agreement 09-022.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.