Article Text

Abstract
Objective We assessed whether the bicarbonate-rich mineral water Staatl. Fachingen STILL is superior over conventional mineral water in relieving heartburn.
Design Multicentre, double-blind, randomised, placebo-controlled trial STOMACH STILL in adult patients with frequent heartburn episodes since ≥6 months and without moderate/severe reflux oesophagitis. Patients drank 1.5 L/day verum or placebo over the course of the day for 6 weeks. Primary endpoint was the percentage of patients with reduction of ≥5 points in the Reflux Disease Questionnaire (RDQ) score for ‘heartburn’. Secondary endpoints included symptom reduction (RDQ), health-related quality of life (HRQOL, Quality of Life in Reflux and Dyspepsia (QOLRAD)), intake of rescue medication and safety/tolerability.
Results Of 148 randomised patients (verum: n=73, placebo: n=75), 143 completed the trial. Responder rates were 84.72% in the verum and 63.51% in the placebo group (p=0.0035, number needed to treat=5). Symptoms improved under verum compared with placebo for the dimension ‘heartburn’ (p=0.0003) and the RDQ total score (p=0.0050). HRQOL improvements under verum compared with placebo were reported for 3 of 5 QOLRAD domains, that is, ‘food/drink problems’ (p=0.0125), ‘emotional distress’ (p=0.0147) and ‘vitality’ (p=0.0393). Mean intake of rescue medication decreased from 0.73 tablets/day at baseline to 0.47 tablets/day in week 6 in the verum group, whereas in the placebo group it remained constant during the trial. Only three patients had treatment-related adverse events (verum: n=1, placebo: n=2).
Conclusion STOMACH STILL is the first controlled clinical trial demonstrating superiority of a mineral water over placebo in relieving heartburn, accompanied by an improved HRQOL.
Trial registration number EudraCT 2017-001100-30.
- acid-related diseases
- anti-reflux therapy
- dyspepsia
- gastric diseases
- gastroesophageal reflux disease
Data availability statement
Data are available on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Bicarbonate-rich mineral waters have been used for decades for symptomatic treatment of gastrointestinal disturbances including heartburn.
Their beneficial effect has not been systematically investigated in a placebo-controlled trial so far.
WHAT THIS STUDY ADDS
STOMACH STILL is the first controlled trial demonstrating superiority of a bicarbonate-rich water over placebo in relieving heartburn.
Symptom relief was accompanied by improved health-related quality of life.
The mineral water was safe and well tolerated.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The mineral water is an alternative treatment option to chemically defined drugs in patients suffering from heartburn that comes along with excellent tolerability.
Introduction
Heartburn is one of the most frequent upper gastrointestinal (GI) tract symptoms1 2 with a variety of causes: whereas many patients have gastro-oesophageal reflux disease (GORD), in some patients, the complaints are only functional without an association between reflux and symptoms or a consequence of other oesophageal diseases.3 4 Of patients with GORD, about 60% have a macroscopically normal mucosa at endoscopy, that is, they have non-erosive oesophageal reflux disease (NERD).5 Reported prevalence estimates of at least weekly GORD symptoms range from 18% to 28% in the USA and from 9% to 26% in Europe.1 Affected patients have a significantly impaired health-related quality of life (HRQoL)—to an extent greater than with disorders like diabetes, arthritis or congestive heart failure. The impaired HRQoL mainly stems from features including disturbed sleep, reduced vitality and pain and has a negative impact on the affected individuals’ productivity in both work and non-work settings.6
According to guidelines for the management of patients with GORD, symptoms including heartburn should be primarily managed by dietary and lifestyle changes—either as initial treatment in mild cases or in combination with pharmacological treatments. However, the evidence base for the effectiveness of lifestyle measures is limited, and management options for symptom relief with proven efficacy are rare.7–9 Recommended chemically defined pharmacological treatments mainly focus on the neutralisation of gastric acid, the reduction of acid secretion and/or the elimination of the acid pocket. These gastric-acid-directed medications include proton pump inhibitors (PPIs) as first-line option besides histamin-2 receptor antagonists, alginates and antacids.
Staatl. Fachingen STILL is a sodium bicarbonate-rich mineral water that has been used for decades for symptomatic treatment of GI disturbances. Its status as a so-called ‘healing water’ qualifies the mineral water as a medicinal product according to German drug law, however, so far, no controlled clinical trial had been available. Preliminary data of Staatl. Fachingen STILL and other bicarbonate-rich mineral waters showed positive effects on heartburn and associated complaints.10–12 The mineral water is expected to exert its beneficial effects by neutralising gastric acid (antacid effect), by accelerating oesophageal clearance and gastric emptying, by increasing gut motility (prokinetic effect) and—as a consequence—by protecting the mucosa of the oesophagus from damage caused by acid reflux.13–15 From a chemical perspective, such mineral waters have the same numerical capacity for neutralising gastric acid as chemically defined antacids.16 The neutralising capacity of the recommended daily dose of 1.5 L mineral water is equivalent to the neutralising capacity of the recommended daily dose of antacids (eg, three tablets/day of Rennie Kautabletten: calcium carbonate/magnesium carbonate).17
The randomised, double-blind, placebo-controlled phase-III trial STOMACH STILL (InveStigation of efficacy and TOlerability of the healing water Staatl. Fachingen STILL in patients for symptoMAtiC treatment of Heartburn in comparison to placebo) was conducted to establish the clinical benefits of Staatl. Fachingen STILL on heartburn (primary objective) and other upper GI complaints as well as HRQoL in a heterogeneous population of patients suffering from heartburn without moderate to severe reflux oesophagitis (Los Angeles (LA) grades B–D). Hence, the clinical trial aimed at providing clinical evidence of symptomatic heartburn relief for a bicarbonate-rich mineral water to extend the therapeutic toolbox for healthcare professionals and patients.
Methods
Study design
This double-blind, randomised, placebo-controlled trial with parallel-group design was conducted from April 2019 to June 2021 in 12 study sites in Germany in accordance with the Declaration of Helsinki (version 2013) and the requirements of the German Medicinal Products Act.
The trial was planned with an adaptive design in two stages to determine the superiority of Staatl. Fachingen Still (verum) over placebo in terms of efficacy for the treatment of heartburn. Placebo was a conventional mineral water with far lower mineralisation than verum. Verum and placebo were visually similar and packed in identical bottles with the same label. Furthermore, both study drugs had a comparable low content of carbonic acid (for composition of study drugs, see online supplemental methods). After screening, the patients went through a run-in phase during which they were advised to drink at least 1.5 L/day of water or other beverages. Only patients with an intake of at least 1.5 L/day of liquids on at least 10 days prior to baseline and with a Reflux Disease Questionnaire (RDQ) score ≥8 in the dimension ‘heartburn’ considering the last 7 days prior to baseline were eligible for randomisation. Patients were centrally allocated to the lowest yet unassigned random number in a blinded fashion to either the verum or placebo group (ratio 1:1, block randomisation with block size n=4). During the treatment period, each patient received for 42 days (6 weeks) either 1.5 L/day of verum or 1.5 L/day of placebo, both to be drunk over the course of the day. The volume of intake was documented in a diary and controlled by the number of empty/full bottles returned. Rescue medication was provided within the clinical trial (Rennie Kautabletten—calcium carbonate/magnesium carbonate); intake was allowed in case the patient considered the heartburn episode as not tolerable and had to be documented. Patients were advised not to change their general eating habits during the trial. Patients, investigators and trial staff remained blinded for the entirety of the trial duration and data analysis.
Supplemental material
The trial is registered in the EU Clinical Trials Register (EudraCT no. 2017-001100-30) and the German Registry of Clinical Studies (DRKS00016696).
Patients
Adults ≥18 years of age were eligible for the trial, if they had a history of repeatedly occurring episodes of heartburn for at least 6 months (RDQ score ≥8 in the dimension ‘heartburn’). An upper GI endoscopy within 5 years before screening excluded relevant erosive disease (reflux oesophagitis LA classification grades B–D) and other severe GI diseases including malignancies, ulcer, Barrett’s oesophagus and oesophageal varices (see online supplemental methods for inclusion/exclusion criteria and explanation of interval between endoscopy and screening). Eligible patients provided written informed consent before enrolment.
Trial parameters: endpoints
The primary endpoint was based on a responder analysis taking into account the severity and frequency of heartburn. For this purpose, the RDQ, a fully validated and reliable instrument for symptom assessment and treatment response in GORD, was used. Within the RDQ, frequency and severity of a symptom (=RDQ dimension) are assessed via two questions each, rated on 6-point scales ranging from 0 (‘no occurrence’) to 5 (‘daily’/’severe’).18 19 A responder was defined as any patient showing a reduction of at least five points in RDQ score for the dimension ‘heartburn’ after 6 weeks of treatment. The chosen responder definition is based on the previously determined minimal important change for a perceived beneficial effect of 4.6 points in the RDQ score for ‘heartburn’.18 20 Symptom changes (RDQ dimensions ‘heartburn’, ‘regurgitation’ and ‘dyspepsia’) were assessed as secondary endpoints. HRQoL was characterised using the Quality Of Life in Reflux and Dyspepsia (QOLRAD), a validated disease-specific questionnaire evaluating the domains ‘emotional distress’, ‘sleep disturbance’, ‘vitality’, ‘food/drink problems’ and ‘physical/social functioning’.21 22 All parameters were assessed at baseline and after 2, 4 and 6 weeks of treatment. Furthermore, the intake of rescue medication based on diary entries was analysed. Also, patients themselves and investigators rated the satisfaction with treatment at all post-baseline visits using the Treatment Satisfaction Questionnaire for Medication version 923 and a 4-point verbal rating scale, respectively.
For characterisation of safety and tolerability, adverse events (AEs) on general questioning, coded according to the Medical Dictionary for Regulatory Activities V.23.0, were analysed according to frequency and severity (mild, moderate, severe).
All analyses presented were prespecified in the trial protocol.
Adaptive design and sample size
Data available in literature did not allow for a reliable sample size estimation and thus, a two-stage approach with interim analysis was planned.24 Appropriate type-I error rates and decision boundaries for the interim (α1=0.0233) and the final analysis (cα=0.00870) were specified to assure control of the global type-I error rate α=0.050.
In a one-arm pilot study (EudraCT no. 2013-001256-36) in 56 patients of a comparable population treated with verum over 6 weeks, a responder rate of ~85% was calculated, and a placebo responder rate of 60% was assumed. A local type-I error rate of 0.0233 was applied at the interim analysis for the primary endpoint yielding a power of 83% for an intended sample size of 130 patients. Thus, considering a drop-out rate of about 15%, 150 patients were planned to be randomised in the first stage. Sample sizes were calculated using the software ADDPLAN, V.6.1.1 (swMATH, Karlsruhe, Germany).
Statistical methods
Efficacy was analysed for all randomised patients who received the study drug at least once, and who provided any postbaseline data for the RDQ score used for responder analysis, and who did not violate against inclusion criteria (full-analysis set, FAS).
For the primary endpoint (responder rate), a two-sided χ2 test (global α=0.05) was used to test for superiority of verum over placebo. Missing data were not imputed; patients without endpoint assessment at study end were regarded as non-responders. Secondary parameters were analysed descriptively. To exploratively assess the treatment effect regarding symptom and HRQoL improvement, an analysis of covariance with baseline as covariate was applied (p<0.05 considered as statistically significant). Safety was analysed descriptively for all randomised patients who received the study drug at least once (safety analysis set, SAS). Statistical analysis was carried out using the software SAS Analyst Pro, V.9.2/0.4 (SAS Institute).
Results
Patients
Figure 1 displays the disposition of patients in the trial. Overall, 148 of 175 screened patients (verum: 73, placebo: 75) were randomised. Treatment compliance was high in the trial population and comparable between the treatment groups: 77.4% of patients (verum: 80.6%, placebo: 74.3%) documented an intake of at least 1.5 L of the study drug on each study day. Five patients (3.4%) did not complete the trial.
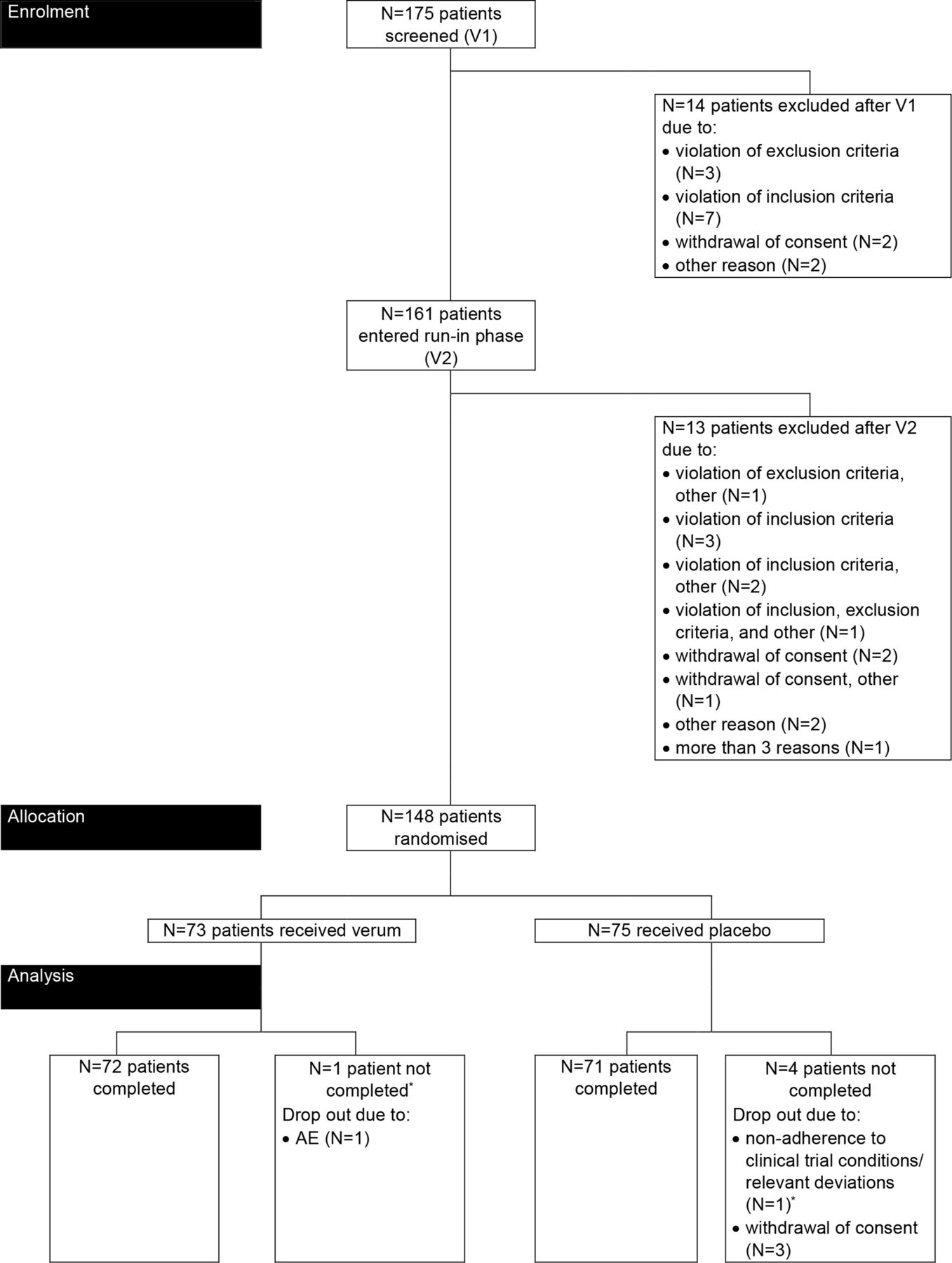
Disposition of patients. *Patient excluded from the FAS due to missing documentation of RDQ score at postbaseline visits V3-V5. AE, adverse event; FAS, full-analysis set; RDQ, Reflux Disease Questionnaire; V, visit.
The baseline characteristics of all patients included in the FAS are depicted in table 1. The patients were all Caucasian, mostly female (66.4%) and had an age between 22 and 81 years; the majority of patients was aged between 51 and 64 years (41.1%). Both treatment groups did not relevantly differ from each other in demographic and anthropometric characteristics.
Patient characteristics (full-analysis set, N=146)*
Primary endpoint: responder rate
In the verum group, 61 out of 72 patients (84.7%) were responders after 6 weeks of treatment, that is, had a reduction of at least 5 points in the RDQ score for the dimension ‘heartburn’. In the placebo group, only 47 out of 74 patients (63.5%) responded to treatment. Hence, a placebo-corrected treatment effect of more than 20% was detected (number needed to treat (NNT) = 5). The primary objective was met: the p value of the χ2 test for the primary evaluation of the RDQ response was p=0.0035, demonstrating superiority of verum over placebo treatment.
Notably, the interim analysis, performed with 144 patients in the FAS, already revealed superiority of verum over placebo (p=0.0034). Consequently, the trial was stopped without the need to proceed to the second stage, and the analysis presented here was then conducted including two additional subjects, who had been included while the interim analysis was done.
Symptom reduction (RDQ)
The RDQ items ‘heartburn’, ‘regurgitation’ and ‘dyspepsia’ as well as the total RDQ score at baseline and after 6 weeks of treatment are summarised in table 2, and the time course of the RDQ item ‘heartburn’ is displayed in figure 2. Baseline scores for ‘heartburn’ and ‘dyspepsia’ were slightly higher in the verum compared with the placebo group. Already after 14 days of treatment, all 4 RDQ scores were clearly reduced, that is, symptoms were improved. Within the last 4 weeks of treatment, the RDQ scores decreased further, although to a smaller degree than in the first 2 weeks. The absolute reductions of all scores from baseline after 6 weeks of treatment were larger in the verum group than in the placebo group, with the greatest difference observed for the dimension ‘heartburn’.

Time course of RDQ score for ‘heartburn’ (full-analysis set, N=146)* Shown are arithmetic mean values with standard deviation. *The full-analysis set includes all randomised patients who received the study drug at least once, and who provided any postbaseline data for the RDQ score used for responder analysis, and who did not violate against inclusion criteria. RDQ, Reflux Disease Questionnaire.
RDQ scores at baseline and end of treatment (full-analysis set, N=146)*
Differences between verum and placebo group reached statistical significance for ‘heartburn’ (p=0.0003) and the total RDQ score (p=0.0050) (table 2). The breakdown of results according to frequency and severity of symptoms revealed that the treatment with verum reduced both characteristics of all three dimensions, although the observed positive effect was larger on the frequency than on the severity of symptoms (online supplemental table 1).
HRQoL (QOLRAD)
The five QOLRAD domains at baseline and after 6 weeks of treatment are summarised in table 3. Baseline scores were either only slightly lower (emotional distress, food/drink problems, vitality) in the verum compared with the placebo group or quite similar (physical/social functioning, sleep disturbance) in both groups. Already after 14 days of treatment, the domain scores were clearly increased, that is, HRQoL impairment was reduced (data not shown). Scores slightly increased further during the last 4 weeks of treatment. The absolute increases of all scores from baseline after 6 weeks of treatment were larger in the verum group than in the placebo group, with the following rank order of placebo-verum comparisons: food/drink problems (p=0.0125) > emotional distress (p=0.0147) > vitality (p=0.0393) > sleep disturbance > physical/social functioning (both p>0.05).
QOLRAD scores at baseline and end of treatment (full-analysis set, N=146)*
Rescue medication
During the baseline interval (comprising the run-in period), the daily average number of tablets of rescue medication (mean±SD) was slightly higher in the verum (0.73±1.15 tablets/day) than in the placebo group (0.56±0.85 tablets/day), which is in accordance with the slightly higher baseline RDQ scores in the verum group. In the verum group, the mean daily use of rescue medication decreased over time to 0.47±1.13 tablets/day in week 6. In contrast, in the placebo group, intake of rescue medication remained nearly constant throughout the trial (0.60±1.44 tablets/day in week 6).
Treatment satisfaction
The patients rated their satisfaction with treatment in the domains ‘effectiveness’, ‘convenience’ and ‘global satisfaction’. The scores for ‘effectiveness’ and ‘global satisfaction’ at all postbaseline visits were higher (= greater degree of satisfaction) in the verum compared with the placebo group. Whereas the degree of treatment satisfaction clearly increased further from week 2 to week 6 in the verum group, it remained nearly constant in the placebo group. The scores for ‘convenience’ were comparably high in both treatment groups and remained constant over time (table 4).
Patient-rated treatment satisfaction post baseline—TSQM-9 (full-analysis set, N=146)*
In accordance with the results of the patient-rated satisfaction with treatment, the investigator assessed the effectiveness of verum at all postbaseline visits as far better than the effectiveness of placebo. Both treatments were assessed with ‘very good’ or ‘good’ tolerability in the majority of the overall population, however, the investigator assessed the tolerability of verum at all post-baseline visits as better than the tolerability of placebo (figure 3).

{kind=link}
{kind=link}
{kind=link}
Investigator-assessed effectiveness (A) and tolerability (B) of treatment post baseline—Verbal Rating Scale (full-analysis set, N=146)*. *The full-analysis set includes all randomised patients who received the study drug at least once, and who provided any postbaseline data for the RDQ score used for responder analysis, and who did not violate against inclusion criteria. N/n, number of patients; RDQ, Reflux Disease Questionnaire.
Adverse events
The summary of AEs per treatment is provided in table 5. The overall incidence of AEs after study drug administration was low (39 out of 148 patients, 26.4%) and comparable between both treatment groups. Only 8 out of 91 AEs (8.79%) were assessed as related to the study drug—all of mild intensity. The related AEs concerned the system organ classes ‘GI disorders’ (verum: 1 case, placebo: 7 cases) and ‘respiratory, thoracic and mediastinal disorders’ (placebo: 1 case ‘throat irritation’).
Adverse events (AEs) per treatment (safety analysis set, N=148)*
Discussion
The present clinical trial represents the first randomised, placebo-controlled, double-blind trial that provides convincing evidence for efficacy of a mineral water in the treatment of heartburn, thereby improving different dimensions of HRQoL.
The phase-III trial was performed in accordance with international standards, the drop-out rate was low and treatment compliance was high. Only two patients were not randomised after the run-in phase due to violation of the inclusion criterion regarding RDQ score, so that a selection bias after run-in can be excluded. The primary endpoint was thoroughly selected: although no standard diagnostic tool for the assessment of the frequency and severity of heartburn is currently available, validated symptom-based patient questionnaires such as the RDQ are regarded as reliable instruments for the assessment of efficacy in clinical trials.19 25–27 Based on the previously determined minimal important change for a perceived beneficial effect of 4.6 points in the RDQ score for ‘heartburn’,18 a responder was defined as a patient with a reduction of at least 5 points in this RDQ dimension score after 6 weeks of treatment. The trial population is highly representative for the overall population of affected individuals with heartburn: according to a large UK database study including 7159 patients, the age distribution of the trial population is largely consistent with the age distribution of patients with GERD in real life.28 Two-thirds of trial participants were female, which is in line with more frequent reports of NERD and reflux symptoms in women compared with men.29
The primary endpoint of the clinical trial was clearly met, demonstrating superiority of verum over placebo in heartburn relief. The detected placebo-corrected treatment effect amounted to >20%, relating to an NNT=5. In comparison, a placebo-corrected effect of 27% was found in clinical trials with the first-line treatment option of PPIs for heartburn relief in patients with NERD (NNT=3.7).30 Consequently, the herein found 20% represent a clinically relevant effect size for a medicinal product with excellent tolerability—especially given the heterogeneous trial population with respect to the underlying cause of the symptom ‘heartburn’.
The reported range of placebo response rates in randomised-controlled trials in GI disorders is broad and can be as high as 47% in trials involving patients with GORD31 and even 73% in trials involving patients with functional dyspepsia.32 The observed placebo response rate of ~60% in the present trial is within that reported range. Furthermore, the consumption of mineral water (1.5 L/day) itself—including the placebo water—may have had a certain symptom relieving effect in the present trial.
The results obtained for all secondary parameters support the primary result: the extent of symptom score reduction with respect to frequency and severity after 6 weeks of treatment exceeded the previously determined minimal important changes for a beneficial treatment effect with respect to all three symptoms,18 although a statistically significant effect over placebo was only apparent for the symptom ‘heartburn’. The smaller effect of the mineral water on the symptoms ‘dyspepsia’ and ‘regurgitation’ is in line with literature data: according to Nocon et al, only the scores for ‘heartburn’ and ‘regurgitation’ are predictive for the assessment of a treatment response, since ‘dyspepsia’ represents a rather non-specific complaint, largely overlapping with reflux symptoms.18 Furthermore, in accordance with the results of the present trial, the symptom ‘regurgitation’ is generally less responsive to acid suppression than ‘heartburn’ in patients with GORD.33 Efficacy of the mineral water in heartburn relief is also reflected by the decreased use of rescue medication in the verum group, while this remained nearly constant throughout the trial in the placebo group.
The reduction in symptom complaints was paralleled by an improved HRQoL: the increases of QOLRAD domain scores after 6 weeks of treatment with verum exceeded the minimal important change of 0.5 points for a perceived beneficial treatment effect in all domains.20 The observed differences between verum and placebo reached statistical significance for the domains ‘emotional distress’, ‘food/drink problems’ and ‘vitality’. These results are largely consistent with the findings from the prospective cohort study ProGERD in which 6215 patients with GORD received the PPI esomeprazole: the extents of improvements in the domain scores after 2 weeks of treatment were comparable to the improvements detected in the present trial. In particular, the highest correlations between the 2-week change in the total RDQ symptom score and the change in the QOLRAD domains in ProGERD were found for ‘food/drink problems’ and the lowest correlations for ‘physical/social functioning’,34 which is in line with the largest effect size for ‘food/drink problems’ and the smallest effect size for ‘physical/social functioning’ in the present trial.
The mineral water was well tolerated with a low AE incidence at placebo level, which is consistent with its known safety profile, as no relevant side effects are known.
A limitation of all studies focusing on symptoms only is that currently no standard diagnostic tool exists for objective assessment of heartburn. Therefore, the results of validated questionnaires based on diary entries by the patients had to be used for endpoint assessment. Furthermore, it could not be fully assured that the two treatment groups were comparable, since oesophageal physiology and factors that may have influenced symptom severity such as psychological comorbidities were not accounted for. Since the included patients had an interval between gastric endoscopy and screening of up to 5 years, patients with moderate to severe reflux oesophagitis (LA grade C/D) might have been mistakenly included. In the ProGERD study (n=2721 completers), however, only 1.6% of patients with NERD and 2.7% of patients with LA grade A/B oesophagitis at baseline had progressed to LA grade C/D oesophagitis after 5 years of follow-up. Furthermore, 4.2% of patients with NERD and 8.1% of patients with LA grade A/B oesophagitis had progressed to confirmed Barrett’s oesophagus after 5 years.35
The trial nevertheless provides the best available evidence because of appropriate internal and external validity and demonstrates a clinically relevant effect size of >20% over placebo with consistent and supportive results in all secondary analyses. Notably, the trial selected patients based on their willingness to drink 1.5 L mineral water per day over 6 weeks, thus, the estimated treatment effect may not necessarily reflect actual impact in real life, if adherence is lower.
In conclusion, the bicarbonate-rich mineral water qualitatively and quantitatively reduced heartburn symptoms in affected adult patients without moderate to severe reflux oesophagitis. The reduction in symptom complaints was paralleled by a relevant improvement in HRQoL and a reduction of the intake of rescue medication. Thus, the results of the placebo-controlled trial STOMACH STILL provide the clinical evidence for a recommendation of the mineral water Staatl. Fachingen STILL in the symptomatic treatment of heartburn. In contrast to chemically defined drugs with their associated long-term side effects, repeated administration of the healing water may result in a continuing and predictable therapeutic effect, along with excellent tolerability.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by the ethics committee of the Thuringian state medical chamber (ethics committee no. 35291/2018/134). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors are grateful to all participating patients and to all involved investigators, especially F. Donath, as well as supporting team members who made this trial possible. The authors thank Staburo GmbH for statistical support and the SocraMetrics GmbH team for data management.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Contributors Concept/trial design: R-SW and BS. Data acquisition: MA. Data analysis/interpretation: JL, MA, JW, R-SW and BS. Writing—first draft: JW. Writing—review/editing: all authors. Final approval: all authors. JL is the guarantor of the article.
Funding The clinical trial was financed by Fachingen Heil- und Mineralbrunnen GmbH, Birlenbach OT Fachingen/Lahn, Germany and conducted by SocraTec R&D GmbH, Oberursel/Erfurt, Germany (CRO).
Competing interests During conduct of the trial and preparation of the manuscript, JL received honoraria for consultancy from Fachingen Heil- und Mineralbrunnen GmbH. MA and JW were employees of SocraTec R&D GmbH. R-SW was an employee of SocraTec R&D GmbH and SocraMetrics GmbH. HW was an employee of Fachingen Heil- und Mineralbrunnen GmbH. BS was cofounder and managing director of SocraTec R&D GmbH and SocraMetrics GmbH.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.