Article Text

Abstract
Background:During the process of tumorigenesis most colon cancer cells acquire resistance to apoptosis. The short chain fatty acid butyrate is well established as an antitumour agent which selectively induces apoptosis in colon cancer cells but not in normal intestinal epithelial cells.
Aims: To analyse the signalling pathway of butyrate induced apoptosis.
Methods: Using Caco-2 cells we focused on the bcl family of proteins, mitochondrial pathway, and caspase signalling cascade involved in butyrate induced apoptosis. Techniques employed included western blots, immunofluorescence, as well as experiments with peptide inhibitors of specific caspases.
Results: Butyrate induced a clear shift of the mitochondrial bcl rheostat towards a proapoptotic constellation, as demonstrated by upregulation of proapoptotic bak accompanied by reduced antiapoptotic bcl-xL levels. This was associated with translocation of cytochrome-c from the mitochondria to the cytosol, resulting in activation of the caspase cascade via caspase-9. Key executioner enzymes were caspases-3 and -1. No effect of butyrate on regulatory proteins of the inhibitor of apoptosis family was observed.
Conclusions: Butyrate induced Caco-2 cell apoptosis via the mitochondrial pathway. Upregulation of bak and translocation of cytochrome-c were upstream of the caspase cascade. Subsequently, this cascade was activated via the formation of an apoptosome.
- SCFA, short chain fatty acid
- AIF, apoptosis inducing factor
- PS, phosphatidyl serine
- ECL, enhanced chemiluminescence
- VDAC, voltage dependent anion channel
- SDS-PAGE, sodium dodecyl sulphate-polyacrylamide gel electrophoresis
- IAP, inhibitor of apoptosis
- XIAP, X linked inhibitor of apoptosis
Statistics from Altmetric.com
- SCFA, short chain fatty acid
- AIF, apoptosis inducing factor
- PS, phosphatidyl serine
- ECL, enhanced chemiluminescence
- VDAC, voltage dependent anion channel
- SDS-PAGE, sodium dodecyl sulphate-polyacrylamide gel electrophoresis
- IAP, inhibitor of apoptosis
- XIAP, X linked inhibitor of apoptosis
Resistance or failure to undergo apoptosis is an integral attribute that characterises the pathogenesis of colorectal tumours.1 An increased understanding of the precise molecular mechanisms underlying this phenomenon could be very beneficial in the development of new agents which restore normal or increased susceptibility to apoptosis. An optimal antitumour drug would be one which selectively induces apoptosis in tumoral but not in normal cells. There is increasing evidence from in vitro and in vivo observations2–5 that the short chain fatty acid (SCFA) butyrate is an agent with such properties. SCFAs are end products of the physiological bacterial fermentation of alimentary fibres in the colonic lumen. It has long been established that SCFAs play a critical role in maintaining homeostatic cell turnover in the colonic epithelium.6 Besides its role as a preferred energy source for normal colonic epithelial cells, butyrate was shown to possess potent anticancer effects on transformed colonocytes. Recent data suggest that the main mechanism responsible for this antitumoral effect of butyrate is induction of apoptosis.7,8 We and others recently reported that butyrate induced Caco-2 cell apoptosis required caspase mediated degradation of repair enzymes and structural proteins.7,8 However, the exact molecular mechanisms by which butyrate activates the apoptotic machinery in tumour cells remain unclear.
Mutations in tumour suppressor genes, such as p53, or in regulatory molecules of the apoptotic signalling machinery play an important role during the process of tumorigenesis of intestinal epithelial cells.9,10 A very common finding in these tumour cells is overexpression of antiapoptotic molecules of the bcl-2 gene family, some of which are under the control of p53.10,11 This steadily increasing family consists of more than 10 proteins with pro- or antiapoptotic properties.12 The different members of the bcl-2-family interact to set a threshold for a cell to undergo apoptosis on specific stimulation. They form heterodimers of an anti- and proapoptotic protein, as well as homodimers, thereby titrating one another's function.12 bcl-2 and bcl-xL are known to strongly inhibit apoptosis at the level of the mitochondrial pathway whereas bax, bak, and bcl-xs are important proapoptotic molecules. It was shown that bax, bak, and bid act directly on the mitochondrial membrane by altering its permeability, probably by forming ion channels.13 This altered outer mitochondrial membrane allows the exit of proapoptotic mitochondrial molecules such as cytochrome-c or apoptosis inducing factor (AIF).13–16 Once in the cytosol, cytochrome-c associates in the presence of ATP with APAF-1 and the proenzyme caspase-9 to form a so-called apoptosome. This in turn results in activation of caspase-9 and further downstream caspases, leading to cell death via apoptosis.17–19
The aim of the present study was to analyse the molecular mechanisms by which butyrate activates the caspase cascade to execute apoptosis in Caco-2 cells. Given the particular importance of apoptosis regulatory proteins of the bcl-2 family during tumorigenesis, in this study we focused on the role of these molecules and the mitochondrial pathway in butyrate induced apoptosis. We observed that butyrate altered the balance of various bcl family members towards a proapoptotic constellation. Subsequent translocation of cytochrome-c from the mitochondria to the cytosol results in activation of the caspase cascade and execution of apoptosis.
MATERIAL AND METHODS
Cell culture and reagents
Caco-2 cells (passages 16–26, ATCC) were cultured at 37°C in a humidified atmosphere of 5% CO2 in minimal Eagle medium supplemented with 5% heat inactivated fetal calf serum (Gibco, Grand Island, New York, USA) and 1% penicillin/streptomycin, as previously described.7
Apoptosis assays
Caco-2 cells were cultured in 24 multiwell plates to subconfluency and stimulated with butyrate (0.01–100 mM) for up to 48 hours. After stimulation with butyrate with or without caspase inhibitors for periods of up to 48 hours, apoptosis was monitored using the annexinV/propidium iodide and TUNEL assay, as previously described.7 Butyrate induced apoptosis was quantified by flow cytometry (FACScan; Becton Dickinson, Heidelberg, Germany). In parallel, morphological characteristics of apoptosis were monitored in butyrate treated Caco-2 cells by confocal and immunofluorescence microscopy, after staining with the DNA dye Hoechst 33342. Expression of phosphatidyl serine (PS) on the outer leaflet of the plasma membrane was monitored by immunofluorescence after binding of FITC labelled annexin-V to PS in the presence of calcium. These studies were performed using a Leica immunofluorescence microscope (Leica, Bernstein, Germany) equipped with a video documentation system. To analyse the role of specific caspases in butyrate induced apoptosis, cells were treated with the following caspase inhibitors: the broad range inhibitor zVAD-fmk (200 μM), the caspase-9 specific inhibitor LEHD-FMK (1–300 μM), the caspase-8 specific inhibitor IETD-FMK (1–300 μM), as well as the caspase-3 specific inhibitor DEVD-FMK (1–200 μM).
Western blots
The effect of butyrate on expression of bcl-2, bcl-xl, bid, bak, and bax, the inhibitors of apoptosis (IAP), cIAP1 and cIAP2, and X linked inhibitor of apoptosis (XIAP), and activation of different caspases were determined by western blots. Butyrate stimulated and unstimulated (control) cell lysates were prepared using an ice cold lysis buffer (50 mM Tris, 150 mM NaCl, 10 mM EDTA, 1% Triton) supplemented with a mixture of protease inhibitors containing antipain, bestatin, chymostatin, leupeptin, pepstatin, phosphoramidon, pefabloc, EDTA, and aprotinin (Boehringer, Mannheim, Germany). Equivalent protein samples were resolved on 8–14% sodium dodecyl sulphate (SDS)-polyacrylamide gels and transferred to nitrocellulose membranes (Bio-Rad, Germany). For immunodetection, membranes were incubated overnight with anticaspase specific antibodies at the following concentrations: anticaspase-1 (1:500), anticaspase-3 (1:1000), anticaspase-8 (1:800), and anticaspase-9 (1:500); antibodies to the bcl-2 family were as follows: anti-bak (1:800), anti-bax (1:700), anti-bcl-2 (1:700), anti-bcl-xL (1:1000), anti-bid-antibody (1:1000; all antibodies from BD-Pharmingen, Heidelberg, Germany), as well as anti-XIAP (1:1000), anti-cIAP1, and anti-cIAP2 (1:600 and 1:1000; all R&D Systems, Wiesbaden, Germany). All antibodies were diluted in Tris buffered saline/Tween 20-1% milk powder. This step was followed by incubation with the corresponding horseradish peroxidase conjugated antibody (antimouse-IgG 1:5000, anti-rabbit-IgG 1:6000; Biosource, Germany). Bands were read by enhanced chemiluminescence (ECL-kit, Amersham, Germany). In addition, mitochondrial expression of bak was analysed after separation of this compartment, as described below.
Cytochrome-c translocation
To monitor the shift in cytochrome-c from the mitochondria to the cytosol, the compartments were separated by ultracentrifugation prior to sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and western blots. Cell pellets were lysed by three cycles of freeze-thawing in a buffer containing 20 mM HEPES/KOH (pH 7.5), 1.5 mM MgCl2, 10 mM KCl, 1 mM NaEDTA, 1 mM EGTA, 1 mM DTT, 0.1 nM PMSF, and 250 mM sucrose, as recently described by Hiura and colleagues.20Thereafter, the cytosolic fraction (S-100 fraction) was separated from the mitochondria by ultracentrifugation at 100 000 g (45 000 rpm) at 4°C for 30 minutes. After SDS-PAGE (15% gel) and transfer onto nitrocellulose membranes, the latter were incubated overnight with an anticytochrome-c antibody (1:500; BD-Pharmingen) followed by a secondary antimouse IgG antibody (1:5000) to perform ECL development, as described above. In parallel, experiments with the same lysates were performed to detect AIF (1:250; Santa Cruz, Heidelberg, Germany).
Immunofluorescence studies
Translocation of cytochrome-c was visualised and monitored by immunofluorescence. Caco-2 cells were grown on glass slides, fixed in 4% ice cold paraformaldehyde after stimulation with butyrate (as above). After intensive rinsing, cells were stained with anticytochrome-c antibody (1:1000; BD-Pharmingen) for two hours at 21°C, followed by a secondary FITC labelled antimouse antibody (1:1000) for 45 minutes at 21°C and photodocumented. Control samples included staining with non-specific isotype mouse IgG, followed by the same secondary antibody and procedure as above. No mitochondrial or cytosol staining was observed in these isotype controls, confirming the high specificity of the primary antibody.
Statistical analysis
Results are reported as mean (SEM) of triplicate samples. Significance was established at 95%, and determined by the Student's ttest for non-paired values and the Mann-Whitney U test for non-parametric values.
RESULTS
Kinetics of butyrate induced Caco-2 cell apoptosis
Incubation of near confluent immature Caco-2 cells with butyrate induced a high rate of apoptosis in a dose and time dependent manner (fig 1A). The proapoptotic effect only occurred above a threshold concentration of 5 mM butyrate. Treatment with butyrate concentrations between 0.1 and 2 mM did not induce Caco-2 cell apoptosis, even after prolonged incubation periods of 48 hours or longer (results not shown). In contrast, butyrate concentrations above 5 mM induced almost complete apoptosis (87 (9)%) at this time interval. Morphological analysis (fig 1B,C) confirmed the typical features of apoptotic cell death in butyrate stimulated Caco-2 cells. Onset of butyrate induced apoptosis was about 16 hours (fig 1A) with—as an early sign—loss of membrane asymmetry, as seen by expression of PS at the outer leaflet of the membrane (fig 1C). Confocal microscopy revealed membrane blebbing in these PS positive cells. The characteristic nuclear changes of apoptotic cell death became more apparent after 20–24 hours of butyrate treatment. Butyrate stimulated Caco-2 cells displayed clear nuclear chromatin condensation and fragmentation resulting in the formation of apoptotic bodies.

Butyrate induced apoptosis in Caco-2 cells. (A) Kinetic analysis revealed that induction of apoptosis occurred after a lag interval of approximately 16 hours after butyrate 20 mM. (B) Confocal microscopy of untreated and butyrate treated Caco-2 cells. The normal ovaloid homogenous surface is totally altered by membrane blebbing and the formation and segregation of apoptotic bodies in butyrate treated Caco-2 cells. (C) In butyrate stimulated cells, expression of phosphatidyl serine (PS) occurred on the outer leaflet of the plasma membrane, an early sign of loss of normal membrane asymmetry. PS expression was visualised by FITC labelled annexin-V, which binds to PS. No PS expression was observed in control cells.
Signalling cascade in butyrate induced Caco-2 cell apoptosis
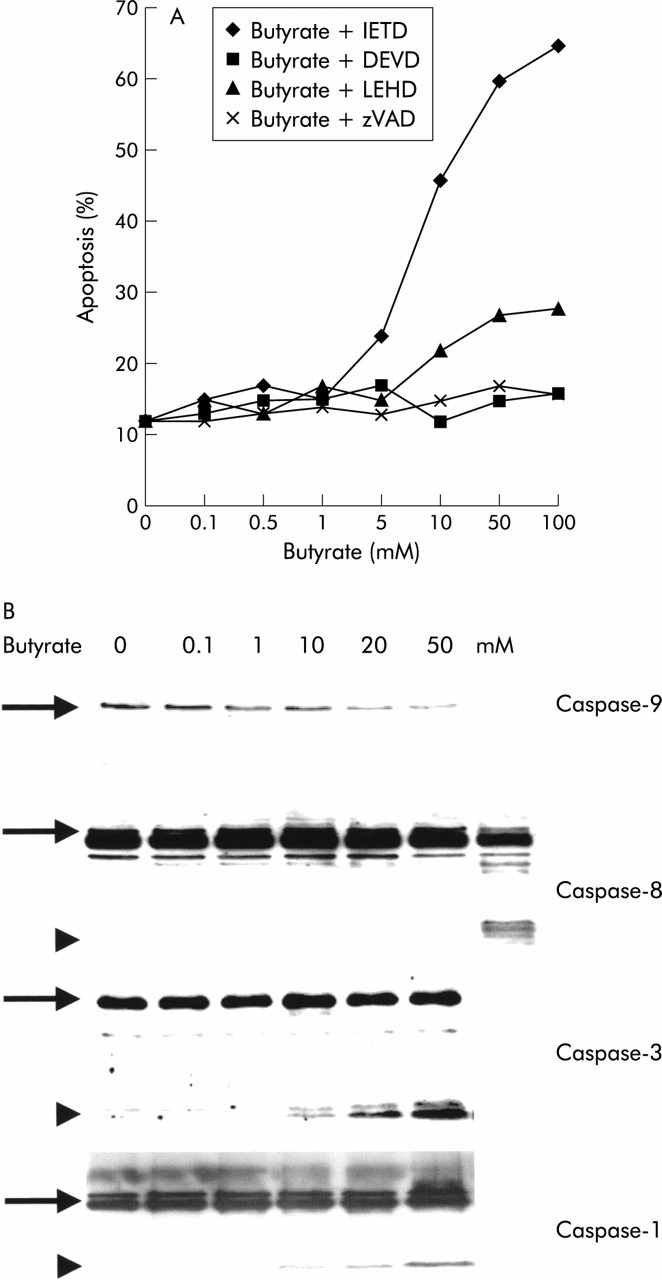
Inhibition of the intracellular apoptosis caspase cascade with the broad range caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluormethyl ketone (zVAD-fmk) completely suppressed the proapoptotic effect of butyrate (fig 2). In order to delineate the molecular events involved, we first examined the “classical caspase cascade” which is activated via a complex of a death receptor, adapter molecules, and caspase-8 (“DISC”).21In response to butyrate, no activation of caspase-8 occurred under all experimental conditions tested, even after prolonged periods of incubation of up to 72 hours (fig 2B). Consistent with this finding was the observation that specific inhibition of caspase-8 with the peptide inhibitor IETD-fmk was also incapable of altering the apoptotic effect of butyrate (48 (9)% versus 46 (11)%). In contrast, we detected clear activation of downstream caspases-3 and -1 in response to butyrate, as demonstrated by western blot (fig 2B). Inhibition of caspase-3 blocked butyrate induced apoptosis (48 (9)% v 12 (6)%; p<0.05), indicating that this enzymes was critical to butyrate induced apoptosis. In contrast, we did not observe any effect of butyrate on expression of any caspases, in contrast with a recent report in a rat colon cancer model.5

Activation of the caspase signalling cascade in butyrate stimulated Caco-2 cells. (A) Use of the broad range caspase inhibitor zVAD-fmk confirmed that butyrate induced apoptosis required a functional caspase cascade. Experiments with highly specific peptide inhibitors directed against caspases-3 (zDEVD-fmk), -8 (IETD-fmk), and -9 (LEHD-fmk) revealed that inhibition of caspase-3 as well as caspase-9 strongly reduced butyrate induced apoptosis, to the point of completely blocking it at certain concentrations. In contrast, inhibition of caspase-8 had no effect. (B) Western blot experiments confirmed the role of the caspase signalling cascade in butyrate induced Caco-2 cell apoptosis. Activation of caspase-9 was suggested by a dose dependent decrease of the band of the inactive proenzyme. For caspases-3 and -1, the active cleaved enzyme was observed in response to butyrate. No effect on caspase-8 was observed. The positive control of caspase-8 activation consisted of TRAIL induced apoptotic Caco-2 cells. One of three representative experiments is shown for each blot.
Several inhibitory proteins have recently been described which block the caspase cascade at specific levels. The molecules XIAP as well as cIAP1 and 2 selectively block caspases-3 and -9.22,23Therefore, we examined expression of XIAP and cIAP1/2 in Caco-2 cells in order to test the hypothesis that butyrate resulted in their downregulation. As shown in fig 3, Caco-2 cells expressed all three IAP proteins. However, even high concentrations of butyrate failed to alter levels of these regulatory proteins.

No effect of butyrate on the antiapoptotic proteins of the inhibitor of apoptosis (IAP) family was observed. Baseline levels of X linked inhibitor of apoptosis (XIAP), cIAP1, and cIAP 2 were high in Caco-2 cells; however, they remained unchanged after stimulation with butyrate. One of three representative experiments is shown for each blot.
Butyrate induced the translocation of mitochondrial cytochrome-c to the cytosol
An important mechanism to activate and stimulate the caspase cascade is via the mitochondrial pathway.19A crucial step in this signalling cascade is the translocation of mitochondrial cytochrome-c into the cytosol. This step allows the formation of a so-called apoptosome, a complex of cytochrome-c, APAF-1, in association with the zymogen caspase-9. This complex then results in the activation of the caspase cascade via cleavage of caspase-9. We observed that butyrate induced a clear shift of cytochrome-c from the mitochondria to the cytosol, as shown by western blot (fig 4A). This efflux was detectable after incubation with butyrate at concentrations of 10–100 mM in a dose dependent manner. Immunofluorescence techniques confirmed the translocation of cytochrome-c into the cytosol in response to butyrate (fig 4B).

Translocation of cytochrome-c from the mitochondrial to the cytosolic compartment in butyrate treated Caco-2 cells. (A) After ultracentrifugation to separate the mitochondrial from the cytoplasmic compartments, a shift of cytochrome-c was observed by western blot in response to butyrate. In contrast, a clear and strong signal for apoptosis inducing factor (AIF) in western blots was only observed in the mitochondrial compartment, without any shift of AIF to the cytosolic compartment, even at high butyrate concentrations. (B) In addition, immunofluorescence analysis confirmed that the perinuclear mitochondrial staining for cytochrome-c in unstimulated cells changed to a diffuse pattern of cytosolic staining in cells after stimulation with butyrate. (C) Inhibition of the caspase cascade with the peptide inhibitor zVAD-fmk (200 μM) did not alter the translocation of cytochrome-c, indicating that this event was independent and upstream of the caspase cascade. One of three representative experiments is shown for each blot.
To test the hypothesis that translocation of cytochrome-c was an initial event and upstream of the caspase cascade, we blocked this signalling cascade using the broad range inhibitor zVAD-fmk (200 μM). At this zVAD-fmk concentration, butyrate induced Caco-2 cell apoptosis was totally blocked (fig 2B). Despite complete inhibition of the caspase signalling cascade, translocation of cytochrome-c occurred in an unaltered way (fig 4C). However, no induction of downstream caspase-3 and Caco-2 cell apoptosis was observed, confirming the necessity of specific caspases to propagate the apoptotic signal.
AIF is another intramitochondrial molecule that can induce the apoptotic signalling machinery by translocation to the cytosol, in a way similar to cytochrome-c.19In contrast with cytochrome-c, we observed no shift in AIF from the mitochondrial fraction to the cytosol in response to butyrate (fig 4B). All AIF remained in the mitochondrial compartment.
We then set out to confirm that translocation of cytochrome-c is involved in activation of the caspase cascade via caspase-9. Specific inhibition of caspase-9 with the peptide inhibitor LEHD-fmk resulted in a dose dependent block of induction of apoptosis, as shown in fig 2A. Western blot analysis confirmed that caspase-9 was activated after stimulation with butyrate (fig 2B).
Effect of butyrate on the bcl-2 gene family
There is increasing evidence that the bcl gene family plays a key role in regulating the release of mitochondrial cytochrome-c. Various antiapoptotic members, such as bcl-2 or bcl-xL, are thought to protect a cell against the translocation of cytochrome-c whereas proapoptotic molecules, such as bax, bak, and bid, promote the efflux of cytochrome-c. In the present study, we therefore examined whether butyrate altered the balance of these pro- and antiapoptotic molecules in Caco-2 cells. Western blot analysis showed marked downregulation of bcl-xL, even at butyrate concentrations as low as 1 mM, whereas no effect on bcl-2 protein occurred even despite maximal concentrations (fig 5). No effect on proapoptotic bax or bid molecules was observed in response to butyrate. On the other hand, bak was strongly induced in a dose dependent manner above a threshold concentration of 5–10 mM butyrate. After separation of the cytosolic from the mitochondrial compartment, clear upregulation of bak in the mitochondria could be observed in response to butyrate stimulation (fig 5). Further fractional analyses revealed however that there was no shift of bax or bid from the cytosolic to the mitochondrial compartment (data not shown).

Effect of butyrate on the mitochondrial bcl rheostat. Western blot experiments of whole cell lysates showed that unstimulated Caco-2 cells expressed high levels of antiapoptotic bcl-xL and lower bcl-2, as well as high levels of the proapoptotics bid and bax. Mitochondrial expression of bak, which under unstimulated conditions was rather low, was markedly enhanced after stimulation with butyrate. No effect was seen on bid or bax levels. On the other hand, bcl-xL expression was markedly decreased by butyrate, even at concentrations as low as 1 mM. One of three representative experiments is shown for each blot.
DISCUSSION
It is well established that butyrate is a potent inducer of apoptosis in colon carcinoma cells.2–6In the present study, we analysed the signalling sequence in Caco-2 cells undergoing apoptosis after stimulation with butyrate. We observed activation of the caspase cascade after an interval of 12–16 hours, in concert with previous reports.7,8The signalling cascade emerged from the mitochondria resulting in initial activation of caspase-9 and subsequently stimulation of downstream caspases-3 and -1. In contrast, no formation of a DISC occurred in response to butyrate, and caspase-8 was not activated. Specific inhibition of this particular caspase with peptide inhibitors did not alter the apoptotic response of Caco-2 cells to butyrate whereas inhibition of caspases-9 or -3 significantly suppressed Caco-2 cell apoptosis.
During the process of tumorigenesis, most cancer cells acquire resistance to apoptosis.9One mechanism is by overexpressing antiapoptotic proteins, inhibiting the signalling cascade at various levels. Since caspases-3 and -9 are key enzymes in butyrate induced apoptosis, we looked for molecules which selectively block these enzymes. XIAP in particular, as well as cIAP1 and cIAP2, are known to potently block both caspases, thereby inhibiting apoptosis.22,23However, we observed no change in the expression of these molecules after butyrate treatment and no inhibitory effect on butyrate induced apoptosis. These observations indicate that these molecules are not involved in the regulation of Caco-2 cell apoptosis induced by butyrate. Another important mechanism which may confer resistance to apoptosis are changes in the bcl-2 gene rheostat. Krajewska and colleagues24 clearly demonstrated in colon cancers that there is significant upregulation of antiapoptotic bcl-xL concomitant with downregulation of proapoptotic bak resulting in a shift towards an antiapoptotic bcl constellation. Similar to that observation, we found in unstimulated Caco-2 cells antiapoptotic bcl-xL expressed at a high level which was in clear contrast with the non-transformed human intestinal epithelial cells HIEC (our unpublished observations). bcl-2 expression was low in Caco-2 cells as was described in normal intestinal epithelial cells.25 Butyrate treatment decreased bcl-xL expression in a potent and dose dependent manner. This downregulation of bcl-xL occurred at concentrations as low as 1 mM butyrate. However, no induction of Caco-2 cell apoptosis was detected at these doses. These data indicate that although downregulation of antiapoptotic bcl-xL was insufficient to induce Caco-2 cell apoptosis, it might decrease the threshold to undergo apoptosis. Nita and colleagues26 reported that resistance to chemotherapeutic agents could be overcome after antisense treatment that suppresses expression of bcl-xL in DLD1 cancer cells. Similarly, Hirose and colleagues27 found in azoxymethane induced rat colonic adenocarcinomas significant upregulation of bcl-xL but moderate downregulation of bcl-2 in tumour cells. These observations confirm that bcl-xL may play a more important antiapoptotic role than bcl-2 in some colon tumour cells as well as in Caco-2 cells.
In contrast, upregulation of proapoptotic bak was a key step in butyrate induced Caco-2 cell apoptosis. We recently showed that suppression of butyrate induced neosynthesis of bak by the protein synthesis inhibitor cycloheximide completely abolished induction of apoptosis in Caco-2 cells.7 Once bak was upregulated, which takes 12–16 hours, cycloheximide failed to block butyrate induced apoptosis, supporting this observation. In the present study, we were able to add additional evidence affirming the crucial role of bak and the mitochondrial pathway in butyrate induced Caco-2 cell apoptosis. After butyrate stimulation, clear upregulation of bak protein was observed in the mitochondria. In contrast, no effect of butyrate on two other proapoptotic molecules, bax and bid, was detectable nor did we observe translocation of either molecule from the cytosol to the mitochondria. Increased levels of bak are known to allow the exit of cytochrome-c from the mitochondrial to the cytoplasmic compartment.28,29 Three dimensional analyses revealed that some bcl family members, such as bax, form ion channels on homodimerisation,29–31 potentially changing the function of mitochondria. The idea has been fostered that these pores produce leakage of the outer membrane, allowing the exit of cytochrome-c or other molecules, such as AIF.14,30–32 Our data indicate that in Caco-2 cells there must be differential regulation of the efflux of cytochrome-c and AIF. We did not observe any translocation of AIF in response to butyrate whereas there was a clear shift of cytochrome-c with subsequent activation of the caspase cascade. Recently, Shimizu and colleagues29 proposed a new mechanism to explain how members of the bcl gene family regulate the translocation of cytochrome-c and induce apoptosis. Using liposomes, they provided evidence that the mitochondrial porin channel, also called voltage dependent anion channel (VDAC), can serve as a channel for cytochrome-c. A significant efflux of cytochrome-c through VDAC occurred only in the presence of bak or bax. Enhanced VDAC activity was dependent on a functional BH3 region of bak or bax. It was speculated that binding to VDAC via this region induced a conformational change in VDAC, allowing the translocation of cytochrome-c. Interestingly, bcl-xL was a competitive inhibitor of this bak effect on VDAC. In the presence of bcl-xL, cytochrome-c release was dramatically inhibited. This antagonistic effect between bak and bcl-xL was regulated by the competitive binding of either molecule to VDAC as well as their heterodimerisation. These observations are in concert with our data. bcl-xL reduction is necessary but not sufficient to allow translocation of cytochrome-c, with subsequent formation of an apoptosome. After upregulation of bak however VDAC became permeable to cytochrome-c, allowing the subsequent activation of the caspase cascade.
Based on the findings in the present study, we propose the following pathway for the induction of apoptosis by butyrate in colon cancer cells (fig 6). The first and crucial step is alteration of the bcl rheostat, with neosynthesis of mitochondrial bak. Enhanced mitochondrial bak expression allows the efflux of cytochrome-c to the cytosol. Decreased bcl-xL levels further facilitate this effect. After formation of an apoptosome resulting in activation of caspase-9, downstream caspases are activated and apoptosis ensues.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proposed signalling pathway for butyrate induced Caco-2 cell apoptosis. After stimulation with butyrate the mitochondrial bcl rheostat is shifted towards a proapoptotic constellation, with markedly increased mitochondrial bak levels and decreased bcl-xL levels. This shift allows the translocation of cytochrome-c from the mitochondria to the cytosol, allowing the formation of an apoptosome. Thereafter, activation of the caspase cascade via caspase-9 ensues, leading to apoptotic cell death.
Acknowledgments
Supported by grants from the Deutsche Forschungsgemeinschaft (DFG Ru 694, 3-1) and BONFOR to FMR, as well as the Dairy Farmers of Canada and the FRSQ to EGS. We acknowledge the excellent technical assistance of T Rottmann.