Article Text

Abstract
Objectives In chronic liver injury, angiogenesis, the formation of new blood vessels from pre-existing ones, may contribute to progressive hepatic fibrosis and to development of hepatocellular carcinoma. Although hypoxia-induced expression of vascular endothelial growth factor (VEGF) occurs in advanced fibrosis, we hypothesised that inflammation may endorse hepatic angiogenesis already at early stages of fibrosis.
Design Angiogenesis in livers of c57BL/6 mice upon carbon tetrachloride- or bile duct ligation-induced chronic hepatic injury was non-invasively monitored using in vivo contrast-enhanced micro computed tomography (µCT) and ex vivo anatomical µCT after hepatic Microfil perfusion. Functional contributions of monocyte-derived macrophage subsets for angiogenesis were explored by pharmacological inhibition of CCL2 using the Spiegelmer mNOX-E36.
Results Contrast-enhanced in vivo µCT imaging allowed non-invasive monitoring of the close correlation of angiogenesis, reflected by functional hepatic blood vessel expansion, with experimental fibrosis progression. On a cellular level, inflammatory monocyte-derived macrophages massively accumulated in injured livers, colocalised with newly formed vessels in portal tracts and exhibited pro-angiogenic gene profiles including upregulated VEGF and MMP9. Functional in vivo and anatomical ex vivo µCT analyses demonstrated that inhibition of monocyte infiltration by targeting the chemokine CCL2 prevented fibrosis-associated angiogenesis, but not fibrosis progression. Monocyte-derived macrophages primarily fostered sprouting angiogenesis within the portal vein tract. Portal vein diameter as a measure of portal hypertension depended on fibrosis, but not on angiogenesis.
Conclusions Inflammation-associated angiogenesis is promoted by CCL2-dependent monocytes during fibrosis progression. Innovative in vivo µCT methodology can accurately monitor angiogenesis and antiangiogenic therapy effects in experimental liver fibrosis.
- FIBROSIS
- ANGIOGENESIS
- MACROPHAGES
- COMPUTER TOMOGRAPHY
- CHEMOKINES
Statistics from Altmetric.com
Significance of this study
What is already known on this subject?
-
Although millions of people worldwide suffer from liver fibrosis and cirrhosis, non-invasive imaging techniques for quantitatively monitoring the progression of liver fibrosis and potential therapy effects in preclinical or clinical settings are currently limited.
-
In advanced liver fibrosis, angiogenesis has been observed in human patients as well as in experimental animal models for chronic hepatic injury.
-
Although fibrosis-associated angiogenesis is regarded as a promoting factor for the transition from chronic injury to hepatocellular carcinoma, the mechanisms of fibrosis-associated angiogenesis remain largely obscure.
What are the new findings?
-
An innovative non-invasive contrast-enhanced functional in vivo micro-CT approach was established in experimental liver injury models in mice and validated by histological and anatomical ex vivo micro-CT-based microstructural analyses.
-
By using these innovative imaging techniques, we demonstrate that (1) even at the initial stages of liver fibrosis, angiogenesis is induced; (2) fibrosis stage correlates with extent of hepatic neovascularisation; (3) infiltrating bone marrow-derived inflammatory monocytes mediate fibrosis-associated angiogenesis induction; (4) pharmacological inhibition of CCL2 attenuates monocyte infiltration and angiogenesis, but not fibrosis progression; (5) these effects are primarily attributable to changes in the portal vein system; and (6) portal hypertension is rather driven by extracellular collagen deposition than by fibrosis-associated pathological angiogenesis.
How might it impact on clinical practice in the foreseeable future?
-
The establishment of a novel non-invasive imaging technique for monitoring fibrosis-associated angiogenesis in the liver and the identification of CCL2-dependent infiltrating monocytes as the driving force of inflammation-associated hepatic blood vessel expansion during fibrosis progression may advance diagnostics and allow novel therapeutic approaches in chronic liver diseases.
Introduction
Liver disease progression is accompanied by pathological angiogenesis,1–3 which is likely a prerequisite favouring development of hepatocellular carcinoma (HCC). However, although pathological angiogenesis is commonly observed in advanced fibrosis, it is currently unclear if and how angiogenesis and fibrosis are linked during progression of chronic liver diseases.4 Interestingly, antiangiogenic multikinase inhibitors have demonstrated antifibrotic potential in preclinical settings,3 ,5 and pro-inflammatory and pro-angiogenic factors are assumed to favour vessel sprouting, resulting in microstructural vascular changes and an increased intrahepatic vascular resistance.6 Whereas several studies have reported that in late stage liver fibrosis or cirrhosis, hypoxia-induced release of pro-angiogenic factors like vascular endothelial growth factor (VEGF) play an important role in the development of HCC,6 ,7 only few studies have investigated the role of inflammatory cells in mediating angiogenesis associated with HCC,8 ,9 non-alcoholic steatohepatitis10 or cirrhosis.3 Based on the pivotal role that (M2-polarised) macrophages play in tumour angiogenesis,11 we hypothesised that macrophages are key mediators of fibrosis-associated angiogenesis as well.
The macrophage pool of the liver is composed of resident immune cells, traditionally termed Kupffer cells, and infiltrating monocytes. In case of liver injury, bone marrow-derived monocytes massively accumulate in injured liver and differentiate into inflammatory macrophages (iMΦ), dependent on interactions of the chemokine receptor CCR2 with its ligand CCL2.12 ,13 These infiltrating monocytes are characterised by the surface marker Ly-6C (Gr1) and a pro-inflammatory cytokine expression profile,14 ,15 while expression of angiogenic factors has not been systematically addressed to date. We therefore aimed at investigating if iMΦ promote fibrosis-associated angiogenesis and, consequently, if the inhibition of monocyte migration into the liver results in a reduced angiogenic activity in chronically injured livers.
In spite of this, no efficient in vivo imaging techniques are currently established, neither for non-invasively quantifying fibrosis-associated pathological angiogenesis nor for monitoring therapy effects of novel pharmacological treatments in (pre)clinical settings. At present, antiangiogenic treatment effects are usually assessed by quantifying CD31-positive vascular structures using immunohistochemistry, and increased CD31+ areas are also observed in virtually all types of advanced hepatic diseases in liver biopsies from human patients.2 Though useful, this methodology does not allow longitudinal examinations during disease progression or quantifications of functional parameters such as blood volume, blood flow and tissue perfusion. In contrast, due to its high spatial resolution, user-independency and suitability for high-throughput analyses, µCT has become a (pre)clinically relevant diagnostic modality for the non-invasive visualisation and quantification of functional blood vessels.16 The aim of this study was to establish novel µCT-based imaging protocols to characterise the role of CCL2-dependent inflammatory monocytes in mediating pathological angiogenesis during the initiation and progression of liver fibrosis. Alterations in the hepatic relative blood volume (rBV) as well as changes in the microarchitecture of liver blood vessels were visualised and quantified using functional in vivo µCT and anatomical ex vivo µCT, convincingly demonstrating that CCL2-dependent inflammatory monocyte-derived macrophages control angiogenesis in experimental liver fibrogenesis in mice.
Materials and methods
Liver injury models and pharmacological CCL2 inhibition in mice
C57bl/6 wild-type mice were housed in a specific pathogen-free environment under ethical conditions approved by German legal requirements. Chronic liver injury was induced in 8–12-week old C57bl/6 wild-type mice by repetitive carbon tetrachloride (CCl4) administration (0.6 ml/kg body weight) or by surgical ligation of the biliary duct (bile duct ligation; BDL), as described earlier.17 To block CCL2-dependent monocyte migration, mice were treated with PEGylated L-RNA Spiegelmer mNOX-E36 (50 nucleotides long L-RNA oligonucleotide; 5′-GGC-GAC-AUU-GGU-UGG-GCA-UGA-GGC-GAG-GCC-CUU-UGA-UGA-AUC-CGC-GGC-CA-3′, 40 kDa PEG), kindly provided by NOXXON Pharma AG (Berlin, Germany), subcutaneously three times per week at 20 mg/kg body weight. mNOX-E36 binds specifically to murine CCL2 (monocyte chemoattractant protein-1 (MCP-1)) and inhibits its biological effects in vitro and in vivo.12 Liver injury, fibrosis progression and immune cell alterations were assessed as published earlier.12 ,17 Primary hepatocytes were isolated by conventional methodology, all other primary cell types (stellate cells, endothelial cells, Kupffer cells, iMΦ) after collagenase/pronase-perfusion, gradient centrifugation and ultrapure FACS sorting using a BD Aria SORP equipped with an UV laser.18 Details on methodology are provided as online supplementary methods.
In vivo µCT
For contrast-enhanced in vivo µCT imaging, a dual-energy flat-panel µCT scanner was employed (TomoScope 30 s Duo; CT Imaging, Erlangen, Germany). Mice were scanned before and immediately after intravenous injection of 100 µL eXIA160XL (Binitio Biomedical, Ottawa, Canada), an iodine-based blood pool contrast agent optimised for in vivo µCT. Animals were anaesthetised with 1.5% isoflurane in oxygen-enriched air during the entire in vivo imaging process. For each mouse, a dual-energy scan was performed at 41 and 65 kV (at 0.5 and 1 mA), acquiring 2880 projections of size 1032×1024 over 6 min of continuous rotation for each tube. A Feldkamp-type reconstruction algorithm (CT-Imaging, Erlangen, Germany) was implemented with a voxel size of 35×35×35 µm3, including ring artefact correction. The reconstructed data were visualised and analysed with Imalytics Preclinical software (Philips Research, Aachen, Germany).19 After 3D liver segmentation, which was performed by interactively delineating the liver boundaries in 10–20 slices, rBV values were determined based on the mean brightness of the liver after contrast agent injection, a large blood vessel after contrast agent injection (100% rBV) and the liver before contrast agent administration (0% rBV).20 Portal vein diameter was quantified on cross-sectional images in transversal planes 3–4 slices above the junction of the superior mesenteric and splenic veins.21
Ex vivo µCT
After in vivo µCT imaging, mice were intracardially perfused with Microfil (Flow Tech, Carver, Massachusetts, USA), a lead-containing silicone rubber CT contrast agent for high-resolution 3D investigation of the microarchitecture of blood vessels in the liver. Microfil replaces the blood volume and polymerises intravascularly 20 min after application, resulting in vascular casting. Perfusion was performed by direct infusion of Microfil into the left ventricle (after incising the inferior vena cava) at physiological pressures by using a perfusion pump. After Microfil perfusion and solidification of the contrast medium, the liver was excised, formalin-fixed and scanned using a high-resolution SkyScan 1172 µCT system (SkyScan, Kontich, Belgium). Livers were positioned on a computer-controlled rotation platform and scanned 180° around the vertical axis in rotation steps of 0.3° at 100 kV and an electric current of 100 µA, resulting in 640 acquired projections (4000×2096 pixels). Acquisition times for whole livers ranged from 6 to 8 h. Reconstructions of a voxel size of 6×6×6 µm3 were performed using filtered backprojection (Feldkamp-type). The volume data were downsampled to a voxel size of 12×12×12 µm3 for image processing. After 3D volume rendering of reconstructed high-resolution µCT data sets, blood vessel branching and 3D micromorphology of vessels were systematically and semiautomatically analysed using Imalytics Preclinical software. For this approach, five representative vessels were analysed, and the number of blood vessel branches per primary vessel was quantified in total and relatively per rising branching order.
Results
Angiogenesis, but not inflammation, correlates closely with progressive fibrosis in chronic liver injury
In order to delineate the association among progressive liver injury, inflammation, fibrosis and angiogenesis, chronic toxic liver injury was induced by repetitive (twice weekly) injections of CCl4 in c57bl/6 wild-type mice. After 2, 4, 6 and 8 weeks, a progressive liver damage including necrotic areas, characteristic fibrotic bridging, infiltration of CD45+ leucocytes and formation of new CD31+ hepatic blood vessels was observed (figure 1A). Interestingly, the kinetics between these principal features differed considerably: while liver cell injury as reflected by ALT activity in serum remained stably elevated throughout the time-course of repetitive CCl4 injections, the infiltration of CD45+ leucocytes into injured livers was most pronounced at early time-points and slowly decreased over 8 weeks (figure 1B). In contrast, the development of hepatic fibrosis as quantified by Sirius red staining (figure 1A) and hydroxyproline concentrations in liver tissue (figure 1B) progressed almost linearly over time. The formation of blood vessels, as analysed by immunofluorescent staining of CD31+ endothelial cells, was strongly induced in progressive liver injury and largely paralleled the progression of fibrosis (figure 1B). These observations demonstrated that angiogenesis and fibrosis progression closely correlate in experimental liver injury.

Association among chronic liver injury, inflammation, hepatic fibrosis and angiogenesis. (A) Chronic toxic liver injury was induced by repetitive intraperitoneal injections of carbon tetrachloride (CCl4) in c57BL/6 mice, and mice were sacrificed 48 h after the last injection of CCl4. Control mice received corn oil for 6 weeks. Representative H&E staining, Sirius red (fibrotic fibres in red), CD45 immunohistochemistry (leucocytes), and CD31 immunofluorescence (blood vessels) of controls and after 4 or 8 weeks CCl4. (B) Serum ALT activity (liver injury), hepatic hydroxyproline content (collagen deposition), CD45+ cells in liver sections (hepatic inflammation) and area fraction of CD31+ endothelial cells (quantification of neovascularisation). Data are shown as mean±SD (n=15 mice). ***p<0.001, **p<0.01 and *p<0.05 (Student t test).
Functional in vivo µCT imaging allows accurate assessment of fibrosis-associated angiogenesis
In order to quantify hepatic blood vessels during fibrogenesis, we established a contrast-enhanced µCT approach for in vivo imaging. To this end, we visualised the 3D micromorphology of hepatic blood vessels in animals treated repetitively with CCl4 after 2, 4, 6 and 8 weeks and in healthy control animals from longitudinal µCT scan series, and we simultaneously quantified non-invasively the rBV in livers using an iodine-based contrast agent optimised for blood pool imaging.20 Via 2D µCT images in transversal, sagittal and coronal planes as well as via 3D volume renderings, dual-energy flat-panel in vivo µCT enabled the non-invasive visualisation of blood vessels in the liver with a spatial resolution of 35 µm voxel side length (figure 2A). Representative images and movies demonstrated that chronically injured livers showed a significantly higher amount of functional blood vessels as compared with vehicle-treated controls (figure 2A, see online supplementary videos 1 and 2). In line, hepatic rBV values were significantly higher for animals challenged with CCl4 for 6 weeks (25.7±0.9%) than for control animals (19.4±0.6%, p<0.0005) (figure 2B), corroborating that fibrosis progression is paralleled by an increasing hepatic blood volume.
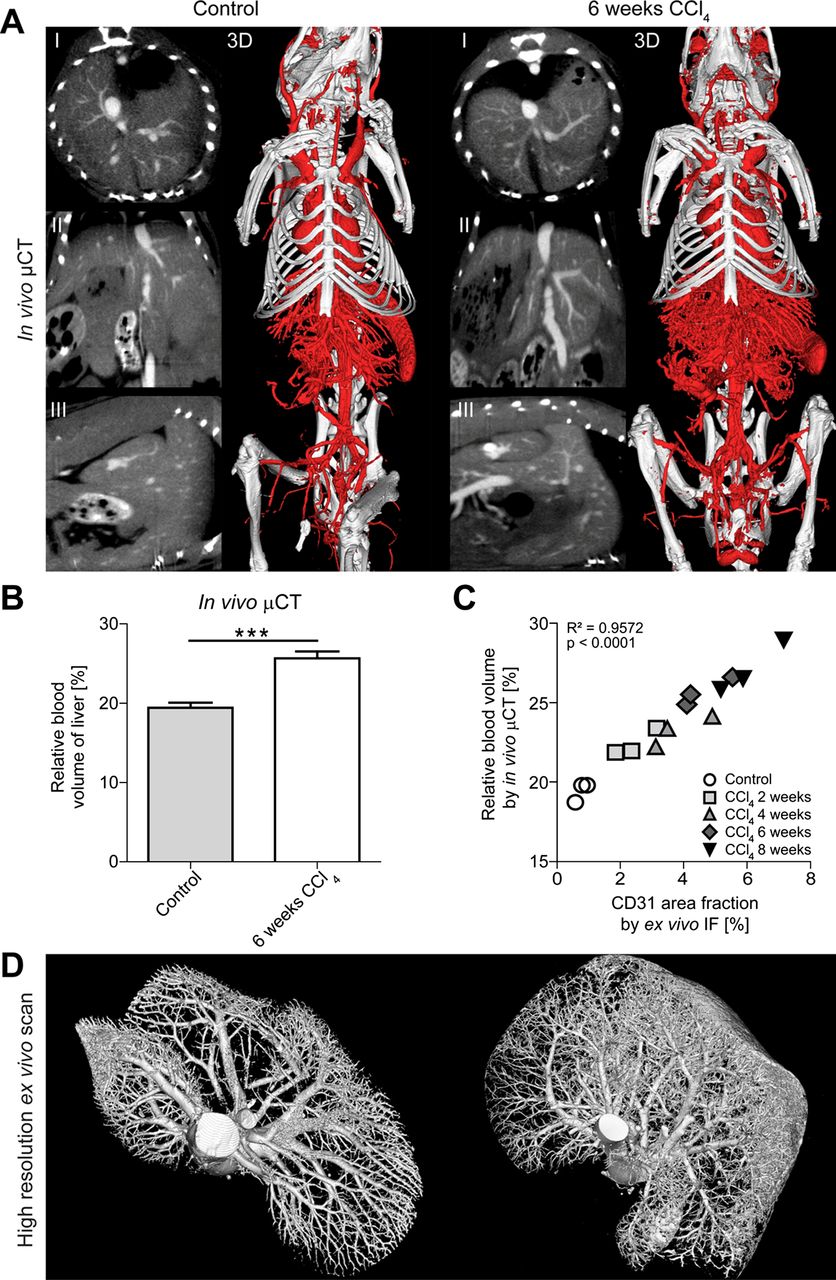
Functional and anatomical µCT imaging of angiogenesis in carbon tetrachloride (CCl4)-induced liver fibrosis. (A) Visualisation of hepatic blood vessels by in vivo µCT using an iodine-based blood pool contrast agent (eXIA 160XL), resulting in a spatial resolution of 35 µm voxel side length (2D cross-sectional images in transversal (I), sagittal (II) and coronal (III) planes, as well as representative pictures of 3D volume renderings). (B) Non-invasive µCT-based quantification of the relative blood volume (rBV) in fibrotic and healthy livers. (C) A highly significant correlation was found between hepatic rBV determined using in vivo µCT and area fraction of CD31 determined using ex vivo immunofluorescence (IF) staining. Data are shown as mean±SD; n=15 mice; ***p<0.001 (Student t test). Correlation analyses were performed by calculating R² (square of Pearson correlation coefficient). (D) High-resolution ex vivo µCT imaging (after perfusion with Microfil, a lead-containing radiopaque contrast agent) enables a detailed 3D examination of vascular microarchitecture of healthy liver (left) and after 6 weeks CCl4 (right). Spatial resolution: 12 µm voxel side length.
To assess the accuracy of in vivo µCT (which considers vessel functionality) versus standard CD31-based immunohistochemistry (which does not consider vessel functionality), rBV values were correlated with the area fraction of CD31+ endothelial cells (figure 2C). Whereas hepatic rBV values determined by in vivo µCT ranged from 22.4±0.9% for animals exposed to CCl4 for 2 weeks to 27.1±1.6% for animals challenged for 8 weeks (control animals: 19.4±0.6%), CD31+ area fractions determined by ex vivo immunohistochemistry ranged from 2.4±0.6% for injured livers after 2 weeks of CCl4 to 6.0±1.0% after 8 weeks of CCl4 (control animals: 0.8±0.2%). A highly significant correlation between rBV values determined using in vivo µCT and ex vivo immunohistochemistry was observed (R²=0.9572, p<0.0001, n=15), indicating that contrast-enhanced in vivo µCT is an accurate means for non-invasively assessing the hepatic blood volume during experimental fibrogenesis.
The spatial resolution of this in vivo µCT-based imaging technique, however, is limited to 35 µm voxel side length, and newly formed blood vessels may partially be smaller. Therefore, we also perfused the animals with the lead-containing vascular casting agent Microfil and performed high-resolution ex vivo µCT scans. Whereas healthy livers showed a proper hierarchic vascular branching system, both for the central and portal vein system, high-resolution ex vivo µCT imaging of fibrotic livers revealed areas with sprouting angiogenesis, in particular in the periphery of chronically injured livers (figure 2D, see online supplementary videos 3 and 4).
In order to exclude model-specific effects related to toxic CCl4-induced damage, we confirmed the close correlation between hepatic fibrosis and angiogenesis in a second mouse model of chronic liver injury (surgical BDL, resulting in progressive cholestatic liver injury). Twenty-one days after BDL or sham operation, mice were imaged using contrast-enhanced in vivo µCT, followed by Microfil perfusion and high-resolution ex vivo µCT imaging, and finally by histological validation. Livers from BDL-treated mice showed severe cholestatic damage with large necrotic areas, bridging fibrosis, leucocyte infiltration and neovascularisation (figure 3A). In line with the results obtained for CCl4, using functional in vivo (figure 3B, see online supplementary videos 5 and 6) and anatomical ex vivo (figure 3C, see online supplementary videos 7 and 8) µCT imaging, a significant increase in the formation of new blood vessels was detected in livers with chronic cholestatic injury. Quantification of liver injury by serum ALT, collagen deposition by hepatic hydroxyproline concentrations and CD45+ leucocytes as well as CD31+ endothelial cells by immunohistochemistry confirmed the presence of injury, inflammation, fibrosis and angiogenesis in BDL-induced liver damage (figure 3D). Accordingly, rBV values were significantly higher in fibrotic (26.3±1.3%) versus control livers (19.6±1.0%, p<0.01) (figure 3D). Collectively, these findings from two independent experimental models for progressive liver fibrosis showed a strong association between liver fibrosis and angiogenesis, and they exemplify that functional in vivo and anatomical ex vivo µCT imaging can be used to monitor fibrosis-associated angiogenesis.

Association between fibrosis and angiogenesis in bile duct ligation (BDL)-induced cholestatic liver injury. Chronic cholestatic liver injury was induced by surgical BDL in c57BL/6 mice. Control animals received sham operations. Mice were imaged and sacrificed 21 days after surgery. (A) H&E staining, Sirius red staining (fibrosis), CD45 immunohistochemistry (inflammation) and CD31 immunofluorescence (blood vessel formation). (B, C) Functional in vivo (B) and morphological high-resolution ex vivo µCT imaging (C) of liver blood vessels from control and from BDL-treated mice (I: transversal, II: sagittal, III: coronal 2D cross-sectional images). Segmented gall bladders (green) illustrate gall bladder hydrops and cholestasis after BDL. (D) Quantification of liver injury via ALT activity in serum, of liver fibrosis by hepatic hydroxyproline levels, of hepatic inflammation by quantifying CD45+ cells, of hepatic blood vessels by determining the CD31 area fraction and of the hepatic relative blood volume determined non-invasively using contrast-enhanced in vivo µCT. Results are shown as mean±SD (n=8 mice). ***p<0.001, **p<0.01 and *p<0.05 (Student t test).
Distinct subsets of hepatic macrophages induce angiogenesis during fibrogenesis
Based on the pivotal role of monocytes and macrophages in the progression of liver inflammation and fibrosis,15 we hypothesised that hepatic macrophages could be the driving force for the formation of new blood vessels during fibrogenesis. Indeed, when mice were challenged repetitively with CCl4 twice per week for up to 8 weeks, a continuous accumulation of F4/80+ macrophages could be observed in injured livers (figure 4A). Upon quantifying the F4/80 area fraction (three different sections per liver, n=15 mice), a massive increase of hepatic macrophages could be detected already at early stages of fibrosis after exposure to CCl4 for 2 weeks (3.9±0.4% vs 0.5±0.2% in healthy livers; p<0.001) (figure 4B). The amount of infiltrated macrophages steadily increased upon prolonged exposure to CCl4 (from 4.7±0.3% after 4 weeks to 6.9±0.8% after 8 weeks). In both models of progressive liver fibrosis, F4/80+ macrophages were localised in close vicinity of newly formed and mainly periportally localised CD31+ blood vessels in chronically injured livers (figure 4C).
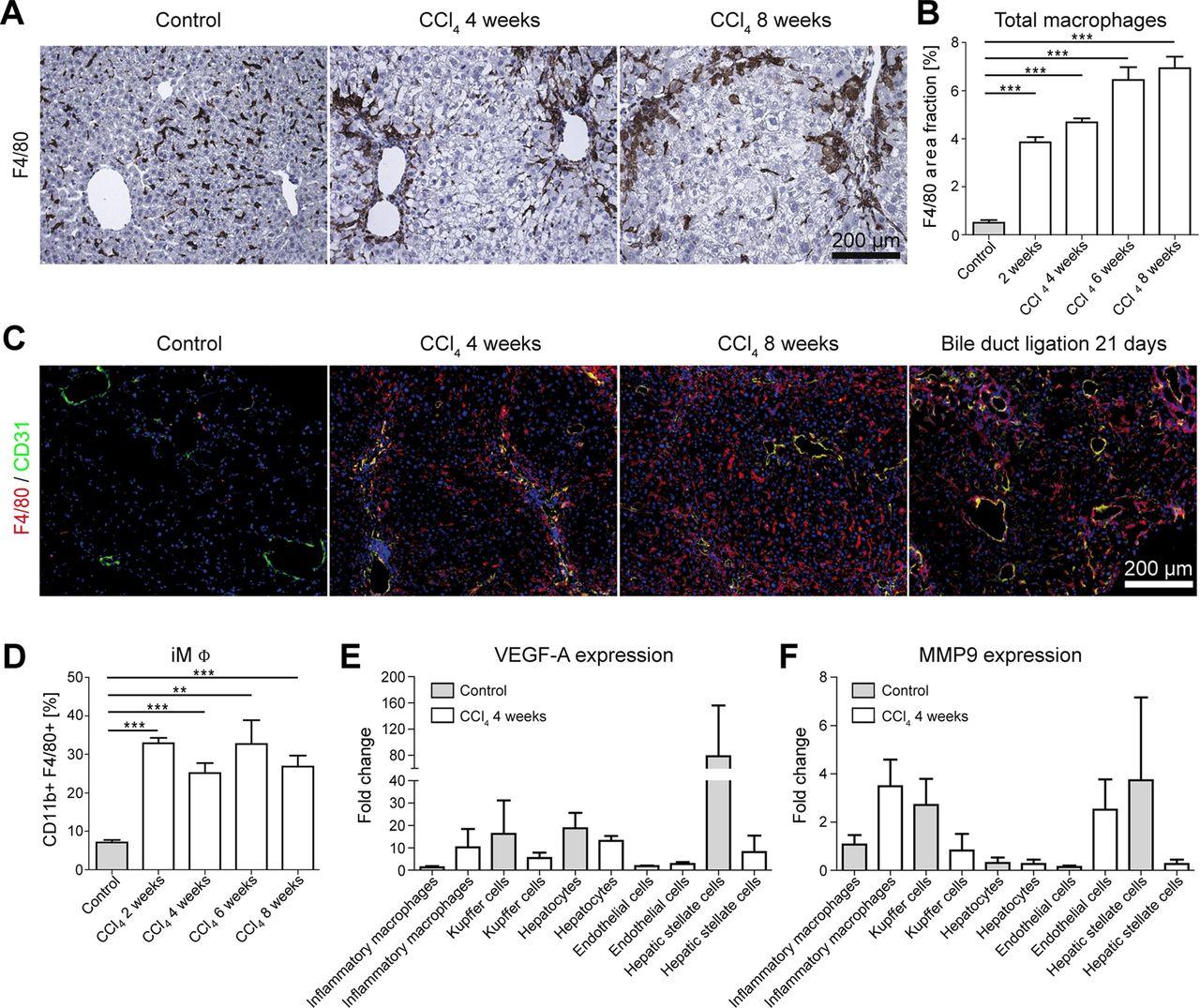
Role of hepatic macrophage subsets in fibrosis-associated angiogenesis. Chronic toxic liver injury was induced by repetitive intraperitoneal administrations of carbon tetrachloride (CCl4) in c57BL/6 mice; control mice received corn oil. Mice were sacrificed 48 h after the last injection of CCl4 (n=6 mice per condition and time-point). (A) Representative microscopy images of F4/80 immunohistochemistry (macrophages). (B) Quantification of total F4/80+ macrophages in sections from control and chronically injured livers. (C) Representative F4/80 (red) and CD31 (green) co-stainings, demonstrating periportal localisation of inflammatory macrophages (iMΦ) and colocalisation of macrophages with newly formed small blood vessels in progressive CCl4 or BDL injury. (D) The relative amount of intrahepatic CD11b+ F4/80+ iMΦ isolated by FACS sorting is early and persistently increased in chronic liver injury. (E, F) Expression of VEGF-A (E) and MMP9 (F) by primary murine iMΦ, Kupffer cells, hepatocytes, endothelial cells and hepatic stellate cells. Cells were isolated from injured (n=12) and control (n=12) livers using FACS sorting, and expression levels were normalised to iMΦ isolated from corn oil-treated control livers. Data are shown as mean±SD. ***p<0.001 and **p<0.01 (Student t test).
A further dissection of iMΦ and Kupffer cells via FACS, based on their differential expression of F4/80 and CD11b, revealed a strong increase of CD11b+F4/80+ inflammatory monocyte-derived macrophages already very early in chronic liver injury, and iMΦ remained the predominant leucocyte population infiltrating throughout the progression of chronic liver diseases (figure 4D).
To determine the pro-angiogenic activity of hepatic macrophage subsets during fibrogenesis, gene expression analyses of purely isolated iMΦ (CD11b+F4/80+), Kupffer cells (defined as CD11blowF4/80++ cells), primary hepatocytes, endothelial cells (CD45−CD146+) and hepatic stellate cells (CD45−UV+) from injured and healthy control livers were performed after FACS-based cell sorting using quantitative real-time PCR. Although Kupffer cells and stellate cells were an important source of angiogenic factors in homeostasis (figure 4E,F), only iMΦ showed a strongly increased expression of pro-angiogenic factors like VEGF-A (10.2-fold higher expression in injured vs control livers) or MMP9 (3.5-fold higher expression in injured vs control livers) upon liver injury. On the contrary, VEGF-A and MMP9 expression was strongly reduced in Kupffer cells, hepatocytes, endothelial cells and hepatic stellate cells in CCl4-treated livers (figure 4E,F). Collectively, these findings strongly indicated that the distinct subset of monocyte-derived iMΦ actively promotes hepatic neovascularisation during fibrogenesis.
Angiogenesis, but not fibrosis progression, is controlled by CCL2-dependent monocyte infiltration
Findings obtained both in mice and in humans demonstrated that the accumulation of inflammatory monocyte-derived macrophages in injured livers is critically controlled by the chemokine CCL2 (also termed MCP-1).15 In order to provide functional evidence that infiltrating monocytes drive angiogenesis in liver fibrosis, we investigated the effect of specific pharmacological inhibition of CCL2 on fibrosis-associated angiogenesis by using the ‘Spiegelmer’ mNOX-E36.12 In fact, administration of mNOX-E36 during CCl4-induced chronic liver injury significantly reduced infiltrating leucocytes, particularly macrophages (as evidenced by F4/80 immunohistochemistry, figure 5A). In line with prior observations,12 mNOX-E36 treatment did neither significantly diminish fibrosis progression nor ALT or AST serum activity in CCl4 injured livers (figure 5A, see online supplementary figure S1). However, blood vessel formation was significantly reduced in mNOX-E36-treated animals (figure 5A,B), and immunofluorescence co-stainings revealed a simultaneous reduction of F4/80+ liver macrophages and CD31+ blood vessels over 2–8 weeks of CCl4 injury in case of mNOX-E36 administration (figure 5A). Importantly, administration of the CCL2 inhibitor mNOX-E36 specifically reduced the amount of CD11b+F4/80+ iMΦ as assessed by FACS analysis from intrahepatic leucocytes (figure 5C), in line with prior findings from our group.12

Effect of pharmacological inhibition of CCL2-dependent inflammatory monocytes on fibrosis-associated angiogenesis. Chronic toxic liver injury was induced by repetitive intraperitoneal administrations of carbon tetrachloride (CCl4) in c57BL/6 mice, and half of these animals received thrice weekly subcutaneous injections of the specific CCL2 inhibitor mNOX-E36, to block the CCL2-dependent infiltration of inflammatory monocytes. Analyses were performed 48 h after the last CCl4 injection. Control mice received corn oil for 6 weeks. (A) Representative H&E staining, Sirius red, F4/80 immunohistochemistry and F4/80-CD31 immunofluorescence co-stainings. (B) Quantification of F4/80+ macrophages and CD31+ blood vessels in livers of chronically injured and mNOX-E36-treated mice. (C) Representative FACS plots and statistical analysis showing the increase of intrahepatic inflammatory macrophages (iMΦ) in chronically injured livers and their significant reduction in mNOX-E36-treated livers. iMΦ were separated from Kupffer cells on the basis of differential expression of F4/80 and CD11b. (D) Liver vascularisation visualised by contrast-enhanced in vivo µCT. (E) Quantification of the relative blood volume in livers and spleens using functional in vivo µCT imaging. Data are shown as mean±SD. ***p<0.001, **p<0.01 and *p<0.05 for comparing CCl4 versus CCl4+mNOX-E36; ###p<0.001, ##p<0.01 and #p<0.05 for comparing CCl4 or CCl4+mNOX-E36 versus corresponding control groups (ie, 6 weeks oil or 6 weeks oil+mNOX-E36) (n=30 mice; Student t test).
In order to confirm and to non-invasively monitor antiangiogenic therapy effects of mNOX-E36 in mice suffering from CCl4-induced liver fibrosis under in vivo hepatic blood flow conditions, functional µCT imaging was performed. Contrast-enhanced in vivo µCT scans demonstrated a significant reduction of fibrosis-associated hepatic blood vessels as a therapeutic effect of mNOX-E36 treatment (figure 5D, see online supplementary video 9). Whereas rBV values rose continuously during the course of CCl4 administration (+15.5%, +20.1%, +32.5% and +39.7% after 2, 4, 6 and 8 weeks, respectively; p<0.01) as compared with control animals, the CCl4-induced increase in rBV was almost completely blocked by mNOX-E36 treatment (figure 5E). At each time-point evaluated, rBV values in the mNOX-E36 therapy group were significantly lower than those in the untreated CCl4 group (−18.8%, −19.4%, −25.9% and −24.4% after 2, 4, 6 and 8 weeks, respectively; p<0.05). Importantly, the effect of CCL2 inhibition was specific to injured livers, because no differences in splenic rBV values were observed in the corresponding animals (figure 5E). Gene expression profiling of livers from mice treated with CCl4 and CCl4+mNOX-E36 for 6 weeks revealed that the reduced numbers of macrophages were reflected by lower expression of macrophage-associated genes such as cytokines, M1/2 proliferation markers and pro-angiogenic factors (see online supplementary figure S1).
In order to exclude that CCL2 or mNOX-E36 itself had direct effects on blood vessel formation, two in vitro experiments were performed. First, primary endothelial cells were isolated from livers of healthy mice, cultured in VEGF-containing medium with or without the presence of mNOX-E36, and either stimulated with CCL2 or left unstimulated (see online supplementary figure S2A,B). Second, using similar culture conditions, an angiogenic sprouting assay with dissected 0.5 mm thin aortic rings of c57bl/6 wild-type mice was performed, analysing the number of sprouts after 10 days (see online supplementary figure S2C,D). Importantly, in both cases of CCL2 stimulation or unstimulated controls, mNOX-E36 treatment did neither affect proliferation and viability of the hepatic endothelium nor the sprouting of aortic rings (see online supplementary figure S2). Taken together, our results demonstrate that the pharmacological inhibition of the chemokine CCL2 effectively blocks fibrosis-associated angiogenesis in chronically injured livers by inhibiting infiltrating CCL2-dependent monocytes.
Macrophage-dependent angiogenesis during chronic liver injury is primarily important for portal vein sprouting
In comparison with functional in vivo µCT, which spatial resolution ends at ∼35 µm, anatomical high-resolution ex vivo µCT imaging, allowing a spatial resolution of ∼12 µm, indicated an even stronger antiangiogenic effect of CCL2 inhibition, especially in the periphery of mNOX-E36-treated livers (figure 6A, see online supplementary video 10). After semiautomatic vessel tree segmentation of the central and portal vein systems, the total number of branching points as well as the number of branching points per rising branching order was quantified (figure 6B). Whereas CCl4-induced toxic liver injury caused a significant increase in branching points in both the central (+59.5%) and portal (+64.0%) vein systems, the CCL2-dependent inhibition of macrophage migration resulted in a clear reduction of newly formed vessels, primarily those associated with the portal vein; as compared with the CCl4 group, 17.6% less branching points of the central vein, and 29.5% less branching points of the portal vein were detected upon CCL2-inhibition (p<0.05 for both; figure 6C). Comparing the distribution of branching points per rising branching order in both tracts, healthy livers demonstrated an almost equal distribution of branching points from the 1st to the 9th order (figure 6D). In progressing liver fibrosis (6 weeks of repetitive CCl4 injections), the formation of new hepatic blood vessels was morphologically linked to a strong increase of branching points in the periphery of both the central and the portal veins: the relative number of 9th order branching points increased significantly (5.6-fold, p<0.0001) and, moreover, 10th and 11th order branching points were newly generated. Conversely, in livers exposed to CCl4 and treated with mNOX-E36, the number of 9th order branching points was again reduced, and this effect was much more prominent in the portal vein (−52.8%) than in the central vein (−25.5%) (p<0.0001; figure 6D). These findings were confirmed using the BDL model (see online supplementary figure S3). Collectively, these highly detailed 3D micromorphological analyses demonstrated that the macrophage-dependent angiogenesis during chronic liver injury is largely confined to portal veins and that pharmacological inhibition of CCL2-mediated inflammatory monocyte infiltration primarily reduces angiogenic vessel sprouting in the portal vein system.

Vascular branching analysis of sprouting angiogenesis in the central and portal vein system in progressive liver fibrosis and upon pharmacological inhibition of CCL2. (A) Representative high-resolution ex vivo µCT images of chronically injured and mNOX-E36-treated livers after systematic Microfil perfusion. (B) Overview and magnification of segmented blood vessels of the liver after semiautomated discrimination between vessels related to the central (blue) or portal vein (red) system. Arrows schematically depict the order of rising branching points along the course of blood vessels, from centre to periphery. (C) µCT-based quantification of the mean total number of branching points in livers from corn oil-, carbon tetrachloride (CCl4)- and CCl4+mNOX-E36-treated mice (all for 6 weeks). Branching points from five representative blood vessels were quantified for both the central and the portal vein system. (D) µCT-based quantification of the percentage of branching points per increasing order (1st to 11th branching order) for livers from corn oil-, CCl4- and CCl4+mNOX-E36-treated mice. Data are shown as mean±SD. ***p<0.001, **p<0.005 and *p<0.05 (Student t test) for comparing CCl4 versus CCl4+mNOX-E36; ###p<0.001, and #p<0.05 for comparing CCl4 or CCl4+mNOX-E36 versus control (Student t test) (D).
Differential impact of fibrosis and angiogenesis on portal hypertension
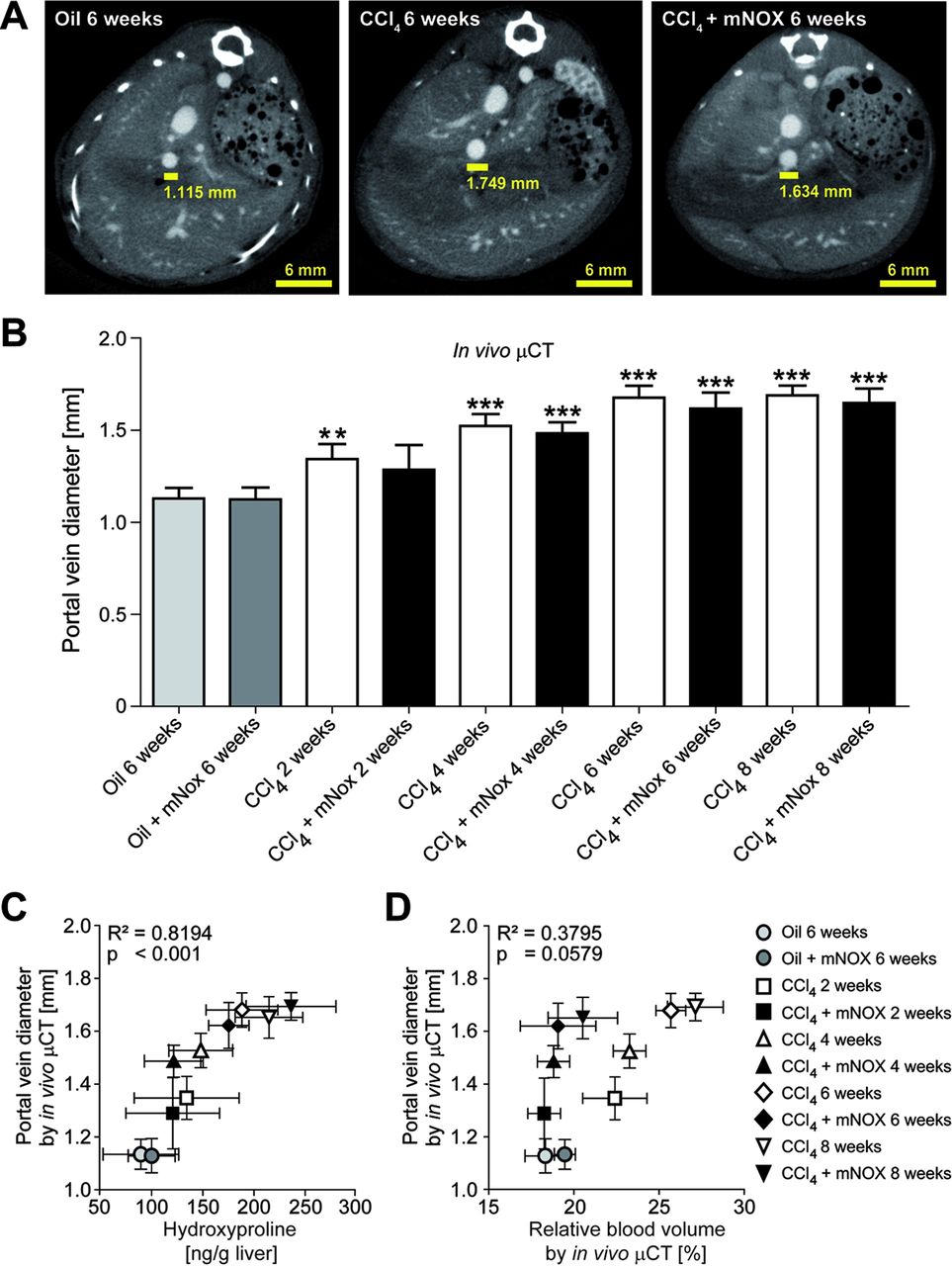
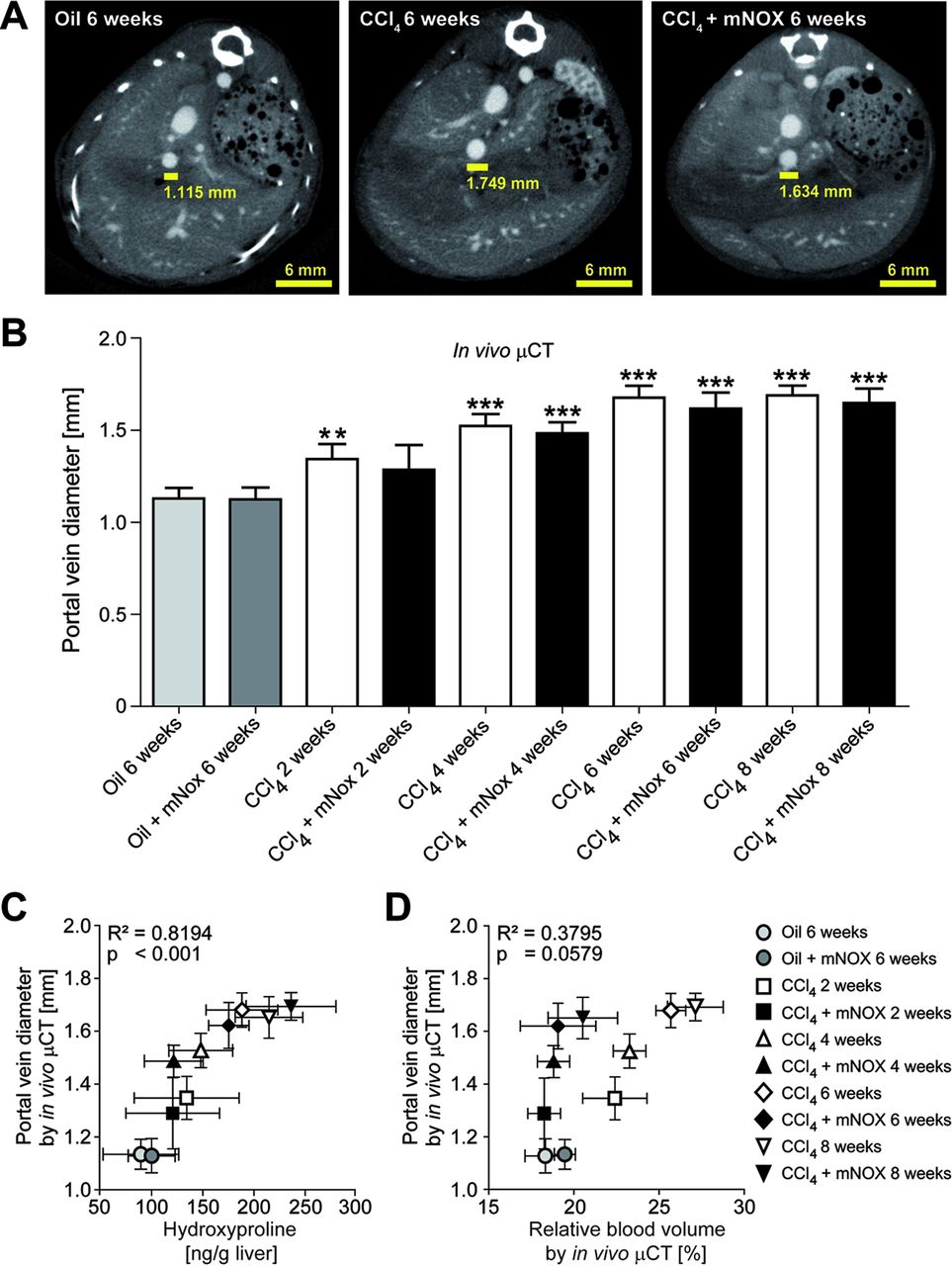
In order to further investigate the association among fibrosis progression, portal vein-related vessel sprouting and portal hypertension, we used contrast-enhanced µCT to quantify the portal vein diameter, as a measure of portal hypertension,21–23 3–4 transversal slices above the junction of the superior mesenteric and splenic veins (figure 7A). Interestingly, portal vein diameter increased significantly during progression of liver fibrosis (eg, 1.68±0.07 mm for animals challenged with CCl4 for 6 weeks vs 1.13±0.06 mm for control animals), but was not significantly influenced by mNOX-E36 treatment (eg, 1.69±0.05 mm for animals treated with CCl4 and mNOX-E36 for 6 weeks; figure 7B). In detail, a highly significant correlation between portal vein diameters determined using in vivo µCT and ex vivo quantified hydroxyproline content was observed (R2=0.8194, p<0.001, n=30; figure 7C). In contrast, no significant correlation was found between portal vein diameters and in vivo quantified rBVs (R²=0.3795, p=0.0579, n=30; figure 7D). These findings strongly indicate that portal hypertension may be more directly caused by the progressive fibrosis itself (extracellular deposition of collagens causing increased liver stiffness) than by pathological fibrosis-associated angiogenesis (sprouting blood vessels causing additional micro-shunts).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In vivo quantification of the portal vein diameter using contrast-enhanced µCT imaging. (A) Cross-sectional images in transversal planes exemplifying the determination of the portal vein diameter 3–4 slices above the junction of the superior mesenteric and splenic veins in control, chronically injured and mNOX-E36-treated livers. (B) Quantification of the portal vein diameter using functional µCT imaging in combination with an iodine-based large molecular weight blood pool contrast agent. (C) A highly significant correlation was found between in vivo quantified portal vein diameters and ex vivo determined hydroxyproline content. (D) No significant correlation was found for the comparison between portal vein diameters and relative blood volumes in carbon tetrachloride (CCl4)-treated, CCl4+mNOX-E36-treated or control mice. Data are shown as mean±SD. ***p<0.001, ** p<0.01 for comparing CCl4 or mNOX-E36 versus corresponding control groups (ie, 6 weeks oil or 6 weeks oil+mNOX-E36) (n=30 mice; Student t test).
Discussion
Chronic hepatic inflammation is regarded as a key requirement for the progression of liver fibrosis, but its role in promoting fibrosis-associated angiogenesis has not yet been elucidated. Moreover, although angiogenesis is commonly observed in patients with hepatic cirrhosis,2 the functional implications of pathological angiogenesis in progressive liver disease have remained largely obscure. Inflammation-associated angiogenesis might contribute to the initiation of liver fibrosis, and in the progression from fibrosis into cirrhosis, and from cirrhosis into HCC.6 ,7 ,9 Thus far, however, studies on the role of angiogenesis in progressive liver disease have been largely restricted to immunohistochemical endpoint analyses. We have here established and employed in vivo and ex vivo µCT-based imaging techniques, alongside conventional molecular methodologies, to demonstrate that (1) even at the initial stages of liver fibrosis, angiogenesis is induced; (2) fibrosis stage correlates with extent of hepatic neovascularisation; (3) infiltrating bone marrow-derived inflammatory monocytes mediate fibrosis-associated angiogenesis induction; (4) pharmacological inhibition of CCL2 attenuates monocyte infiltration and angiogenesis, but not fibrosis progression; (5) these effects are primarily attributable to changes in the portal vein system; and (6) portal hypertension is rather driven by extracellular collagen deposition than by fibrosis-associated pathological angiogenesis.
Prior studies have indicated that hypoxia increases the expression of the prototypic pro-angiogenic factor VEGF, thereby promoting angiogenesis and potentially also the progression from fibrosis to cirrhosis and to HCC.1 ,3 ,7 VEGF enhances endothelial cell proliferation, promotes vessel sprouting and branching, and increases microvessel permeability.24 Besides classical hypoxia-induced and HIF1-mediated VEGF expression, inflammatory cells might also elevate the levels of pro-angiogenic factors in injured livers.10 ,25 In experimental models of cancer progression, infiltrating iMΦ are a major mediator of tumour angiogenesis.26 In line, inflammatory cells migrating into injured livers have been implicated in HCC-associated angiogenesis.8 ,9 Detailed analyses on the molecular link among inflammatory monocyte infiltration, angiogenesis and liver fibrosis progression, however, have not yet been performed to date, which can be at least partially explained by the relative lack of suitable imaging techniques to non-invasively, longitudinally and quantitatively monitor angiogenesis.
The chemokine-dependent accumulation of monocyte-derived macrophages has been identified as an important mechanism for perpetuating hepatic inflammation and promoting fibrogenesis in experimental mouse models as well as in human liver diseases.15 Upon experimental organ injury in mice, the chemokine receptor CCR2 and its ligand CCL2 (MCP-1) promote monocyte subset accumulation in the liver, in particular of Ly6C+ (Gr1+) monocytes. These monocyte-derived macrophages release pro-inflammatory cytokines and can directly activate pro-fibrogenic hepatic stellate cells, the main collagen-producing cells in the liver.15 Our study significantly extends these previous findings by demonstrating that infiltrating CCL2-dependent inflammatory monocytes also provide pro-angiogenic signals, likely via the production and release of VEGF-A and other factors like MMP9.
To test our hypothesis that these infiltrating monocytes are essential in mediating fibrosis-associated angiogenesis, the CCL2/CCR2-dependent migration of inflammatory monocytes into injured livers was inhibited using a novel CCL2 inhibitor, the ‘Spiegelmer’ mNOX-E36.12 Findings from quantitative in vivo and ex vivo µCT imaging of functional liver blood vessels, coupled with FACS and gene expression analyses, revealed that the inhibition of CCL2-dependent inflammatory monocytes by mNOX-E36 significantly reduced the formation of new blood vessels in progressive liver fibrosis. Thus, mNOX-E36, whose human equivalent is currently being evaluated in a phase II clinical trial for the treatment of diabetic nephropathy (http://www.clinicaltrials.gov, NCT01547897), appears to be an attractive therapeutic approach for reducing the inflammation-associated angiogenesis in progressive liver fibrosis. Strikingly, the inhibition of CCL2-dependent monocyte infiltration did not affect liver fibrosis progression per se, in line with prior work from our group.12 This further demonstrates that the inhibition of macrophage-mediated angiogenesis induction did not attenuate the progression from early-to-late stage liver fibrosis, indicating that initially angiogenesis is a consequence of inflammation, rather than a cause for disease progression. However, at later stages, the contribution of pathological angiogenesis to chronic liver disorders is anticipated to become more and more causal, in particular during the transition from cirrhosis to HCC, and the subsequent progression and spread of HCC.
Besides identifying a yet unrecognised cellular and molecular mechanism of fibrosis-associated angiogenesis, our study may also be of interest for developing future imaging strategies in the management of patients with chronic liver disease. The current ‘gold standard’ of fibrosis assessment is needle biopsy of the liver, carrying the risk of bleeding complications as well as sampling errors.27 In the last couple of years, only few ultrasound and MR-based methods have been evaluated for non-invasively measuring liver stiffness.4 ,28 ,29 Some studies reported on the non-invasive quantification of the portal vein diameter via MR or ultrasound as a measure of portal hypertension.21–23 In addition, very few studies described molecular imaging approaches for the in vivo assessment of liver fibrosis via collagen- or elastin-specific contrast agents in experimental settings.30 ,31 However, due to the fact that no antifibrotic drugs have been approved up to date,4 only limited data exist for these imaging approaches for therapy monitoring. In contrast, functional imaging approaches like quantitative contrast-enhanced CT enable the visualisation of liver blood vessels, non-invasive quantification of the hepatic rBV, which correlates significantly with the amount of newly formed blood vessels during fibrosis progression, and quantification of the portal vein diameter, which correlates with degree of portal hypertension. With respect to pathophysiological circulation conditions in fibrotic livers, µCT imaging can assess microstructural and functional changes in the hepatic vascular network and provides in addition the unique opportunity to monitor antiangiogenic therapy effects. We show here that µCT imaging is highly useful for quantitatively assessing fibrosis-associated angiogenesis, and for monitoring anti-inflammatory and antiangiogenic therapy effects in progressive liver disease, indicating that anatomical and functional contrast-enhanced CT imaging might hold great potential for facilitating the bench-to-bedside translation of novel targeted treatments and therapeutic interventions.
Acknowledgments
The authors thank Aline Roggenkamp, Carmen Tag and Sibille Sauer-Lehnen for excellent technical assistance and NOXXON Pharma AG (Berlin, Germany) for providing mNOX-E36.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online video 1
- Data supplement 4 - Online video 10
- Data supplement 5 - Online video 2
- Data supplement 6 - Online video 3
- Data supplement 7 - Online video 4
- Data supplement 8 - Online video 5
- Data supplement 9 - Online video 6
- Data supplement 10 - Online video 7
- Data supplement 11 - Online video 8
- Data supplement 12 - Online video 9
Footnotes
JE and MB contributed equally.
-
Contributors JE, MB, XW, FG, VF and DM: performed experiments, acquired data and analysed results; CB and DE: contributed methodology for CCL2 inhibition; KH: performed immunohistochemistry and analysed results; TLu, FK and CT: helped in data interpretation and provided important intellectual content; JE, MB, TL and FT: designed the study, analysed data and wrote the manuscript.
-
Funding This work was supported by German Research Foundation (DFG; SFB/TRR57, TA434/2-1, EH412/1-1 and LA2937/1-2), Interdisciplinary Center for Clinical Research (IZKF Aachen) and European Research Council (ERC-StG-309495-NeoNaNo).
-
Competing interests Dirk Eulberg is an employee of Noxxon Pharma AG. All other authors have nothing to disclose.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement We agree to share all primary data and protocols employed in this study.